⚡️ 【Nat. Biomed. Eng.】颠覆性突破:AI预测蛋白质丰度,精确度超87%!单细胞分析成本有望降低90%?
✨文章标题:A pre-trained large generative model for translating single-cell transcriptomes to proteomes ✉️作者: Ka-Chun Wong, Fan Yang & Jianhua Yao 等 🔗链接:https://doi.org/10.1038/s41551-025-01528-z

🔬 从“痛点”到“突破”:为什么细胞世界的“翻译官”如此重要?
在生命科学领域,我们一直追求一个“高分辨率”的视角,想要看清疾病和细胞过程中的每一个分子机制。这就好比我们想看一场精彩的电影,不仅要知道剧本(基因),更要知道演员的表现(蛋白质)。因为,真正驱动细胞生命活动的,是蛋白质。
然而,当前的单细胞蛋白质组学技术却面临着一系列“老大难”问题:覆盖的蛋白质种类有限、检测灵敏度和通量不足、实验成本极其高昂,而且操作复杂。测量一个细胞中的蛋白质,比测量它的基因信息要难上太多。
雪上加霜的是,细胞内的“翻译”过程并不完美。基因信使(mRNA)的丰度,大约只能解释40%到60% 的蛋白质丰度变化。在单个细胞层面,这个挑战被细胞间的转录状态、翻译速率和蛋白质周转差异进一步放大了。这就形成了一个巨大的瓶颈:我们手握海量的单细胞基因信息(scRNA-seq),却缺少最关键的蛋白质信息。
有没有一种方法,可以像自然语言处理(NLP) 中的机器翻译一样,直接将“基因语言”精准地“翻译”成“蛋白质语言”呢?来自顶尖研究机构的科学家们,给出了一个颠覆性的答案:scTranslator——一个预训练的大型生成模型,它能够仅根据单细胞转录组数据,推断出缺失的单细胞蛋白质组。
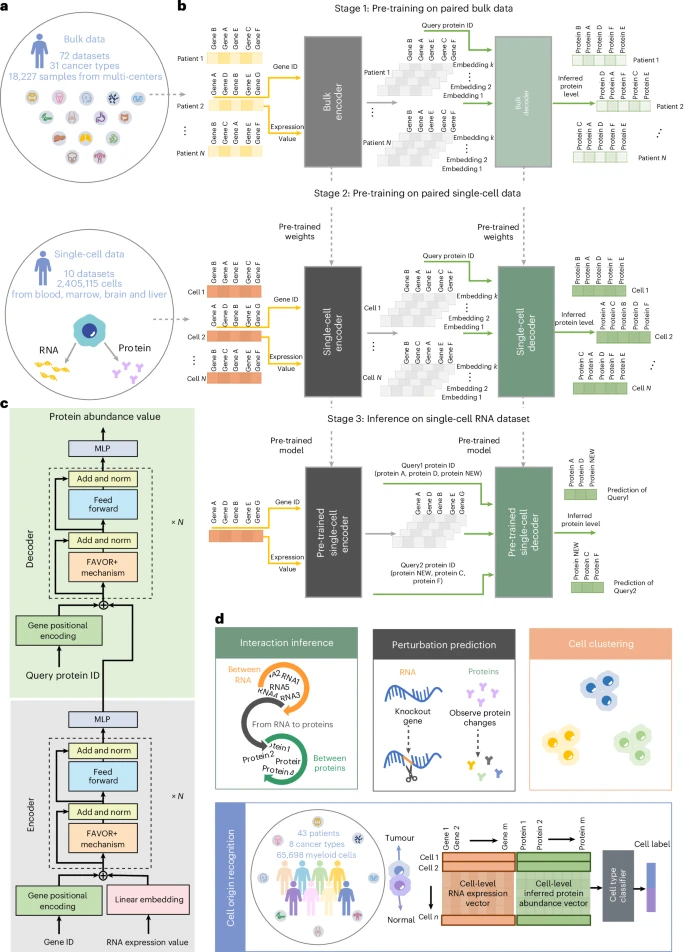
🧠 核心方法与技术细节解密:AI如何成为细胞世界的“中央教义翻译官”
scTranslator的诞生,灵感恰恰来源于生物学的“中央教义”(Central Dogma,即基因信息从DNA流向RNA,再到蛋白质)以及人工智能领域的翻译模型。
我们都知道,Transformer模型是自然语言处理(NLP)领域的“明星”,它擅长理解和生成长序列信息。scTranslator的核心,正是对Transformer架构进行了一次“生物学定制改造”。
1. 大规模“双语”预训练:构建生物学知识的“大词典”
一个优秀的翻译家,需要足够大的知识储备。scTranslator的训练采用了独特的两阶段预训练策略,来构建一个跨越不同层次和条件的“生物学大词典”:
第一阶段(宏观学习): 模型首先在批量(Bulk)数据上进行训练。这些数据来自31种癌症、18,227个患者样本,包含基因和蛋白质的配对信息。批量数据成熟且蛋白质覆盖全面,不仅有细胞表面蛋白质,还有细胞内部蛋白质,帮助模型建立起宏观、全面的基因-蛋白质关系基础。
第二阶段(微观精修): 接着,模型在超过200万个单细胞的配对数据上进行持续学习。这一步让模型能够捕捉到单细胞层面精微的细胞间变异,将从宏观数据中获得的“通用知识”打磨成针对单个细胞的“精细技能”。
2. 独家创新:为基因信息注入“位置感”的GPE模块
在传统的Transformer模型中,每个词语都有一个“位置编码”来指示其在句子中的位置。在细胞世界,哪个“基因”对应哪个“位置”呢?
研究团队创造性地设计了 “重新索引的基因位置编码”(Re-indexed GPE)模块。这就像是给每一个基因赋予了一个独一无二、且带有层级关系的身份ID:
传统的基因ID号庞大而分散,容易造成高内存占用和低效率。GPE模块将所有官方认证的基因(超过75,500个)重新编号为一个连续且有意义的ID,并将其转化为一个可学习的“位置向量”。
这个巧妙的设计,不仅使得模型能够区分来自不同基因的表达值,更重要的是,它通过分配相近的ID给相似的基因,在编码中保留了基因之间的邻近关系,极大地增强了模型对基因互作的理解能力。
3. 挑战长序列:从平方级到线性级的“提速”
单细胞转录组数据动辄包含数万个基因,这远超NLP领域的普通句子长度。如果使用标准的Transformer,其计算复杂度将是序列长度的平方级别,难以承受。
scTranslator通过采用FAVOR+ 机制(Fast Attention Via positive Orthogonal Random features),成功将计算复杂度降至线性级别。这让模型能够更高效地处理长序列的基因和蛋白质数据,捕捉长距离依赖关系,实现了高可扩展性。
4. “一步到位”的解码器:效率飞跃
与传统的Transformer解码器需要一步步预测下一个词语(即“自回归”解码)不同,scTranslator采用了一种 “一次前向操作”的生成式解码策略。这意味着它可以瞬间预测出所有被查询的蛋白质丰度,大幅提升了推断效率,特别适合于大规模、长序列的蛋白质预测。
📈 数据背后的创新与颠覆性分析:为什么说它是一个了不起的成就?
scTranslator的性能,已经不仅仅是“能用”的水平,而是达到了颠覆现有技术瓶颈的程度。
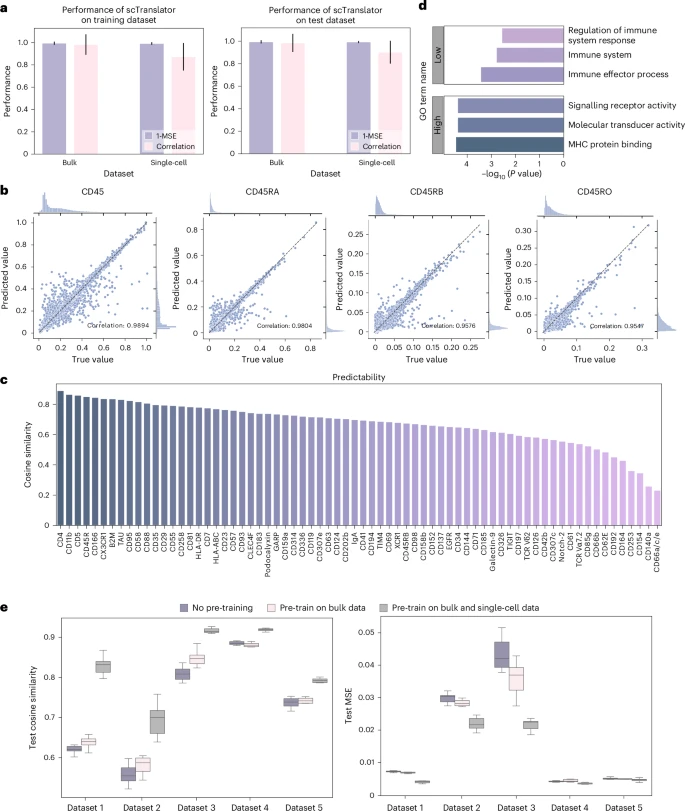
1. 预测精度:在单细胞级别实现了超高保真度
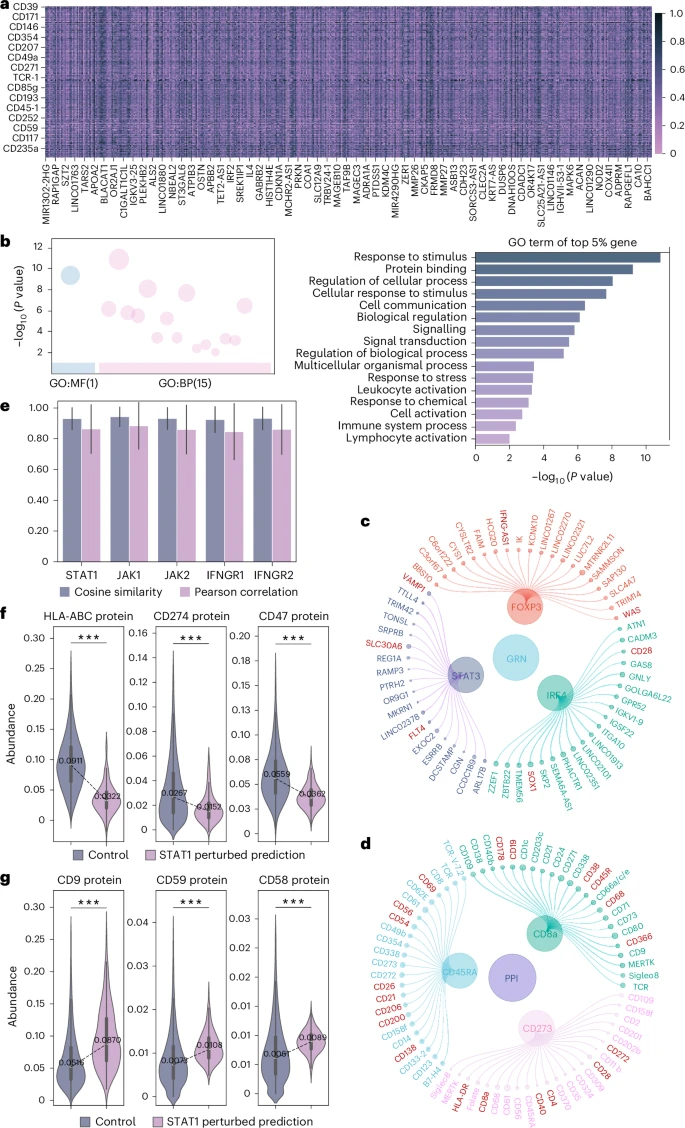
在对看不见的(测试集)单细胞数据进行预测时,scTranslator在余弦相似度(Cosine Similarity) 上达到了超过0.87的成绩,均方误差(MSE)低于 。余弦相似度是衡量预测值与真实值方向一致性的关键指标,0.87意味着模型对蛋白质丰度水平的预测具有极高的保真度。
即使面对CD45及其多个剪接变体(如CD45RA、CD45RB、CD45RO)的复杂情况,模型的预测相关性依然达到了0.95至0.98的惊人水平 ,这表明它能精确区分具有相似功能的蛋白质家族成员。
2. 战胜数据稀缺:在“零星数据”场景中的性能暴增
在真实世界的科研中,我们往往只能获得非常有限的配对数据,即所谓的 “少样本学习”(Few-shot learning) 场景。这是衡量预训练模型泛化能力的终极考验。
在仅使用20个细胞作为训练数据,其他细胞用于测试的极端少样本实验中 ,scTranslator的表现碾压了所有SOTA(State-of-the-art,现有最先进)方法:
相比于SOTA的Seurat,scTranslator在余弦相似度上实现了最低6.6%,最高80.7% 的显著提升 。
这充分证明了其两阶段预训练策略的巨大成功:模型已经从大规模数据中获得了极其丰富的“通用生物学知识”,以至于只需要极少的、零星的新数据,就能迅速适应新的细胞环境并做出高精度的预测 。
此外,在探究模型性能与训练数据量的关系中,研究发现,虽然更多的训练数据有益,但仅需20个细胞,scTranslator就能达到使用90%数据进行训练时80%到95%的性能 。这对于急需低成本、快速启动的单细胞项目来说,无疑是革命性的效率提升。
3. 应对“错位”数据:GPE模块的决定性胜利
在多中心、多技术平台协作的科研中,由于检测基因和蛋白质集的差异,数据往往是**“非对齐”(Un-aligned)的。这要求模型必须能处理在训练集中从未见过的新基因或新蛋白质**。
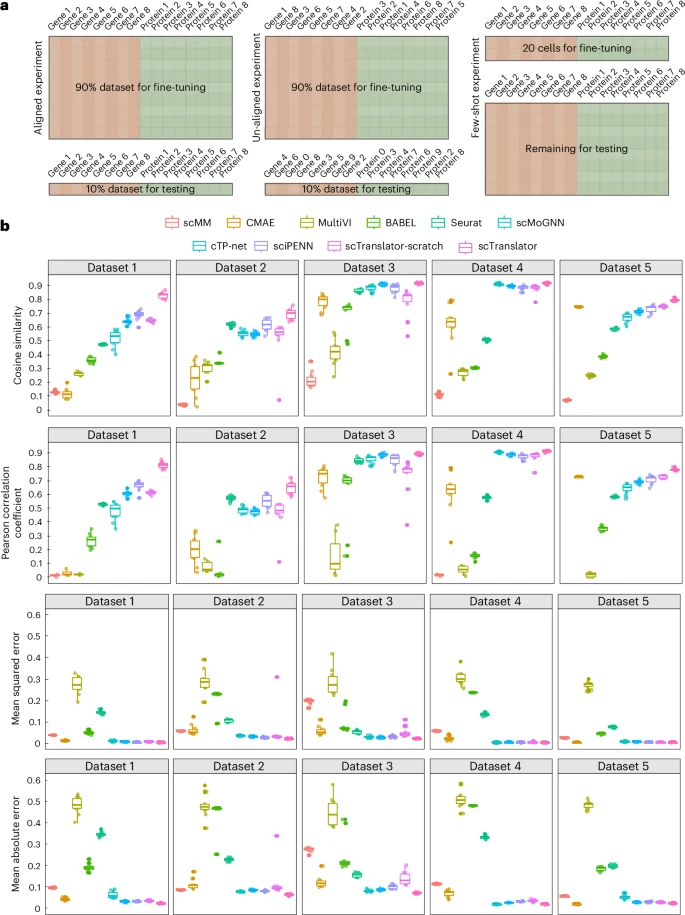
传统的SOTA方法在“非对齐”实验中表现不佳,一半的方法在皮尔逊相关系数(PCC)上甚至未超过0.3 。而得益于其独创的GPE模块,scTranslator在五个测试集上的PCC分数均高达0.793到0.952 ,展现出卓越的鲁棒性。这表明,研究团队定制的Transformer架构在捕获转录组和蛋白质组关系方面具有无可比拟的优势 。
4. 预测扰动效应:媲美真实实验的计算推演
更令人振奋的是,scTranslator能够以计算方式,预测基因扰动(Gene Perturbation)对蛋白质水平的影响 。这极大地降低了体外实验的时间和资源成本。
在模拟干扰IFN-通路基因(如STAT1、JAK1等)的Perturb-CITE-seq实验中,scTranslator的预测蛋白质水平与实际测量的结果在余弦相似度和PCC上均超过0.8 。这意味着,该模型能够在未经过扰动数据训练的情况下,准确地捕捉到基因表达与蛋白质水平之间深层次的调控关系,预测出与文献报道一致的蛋白质上调或下调模式 。
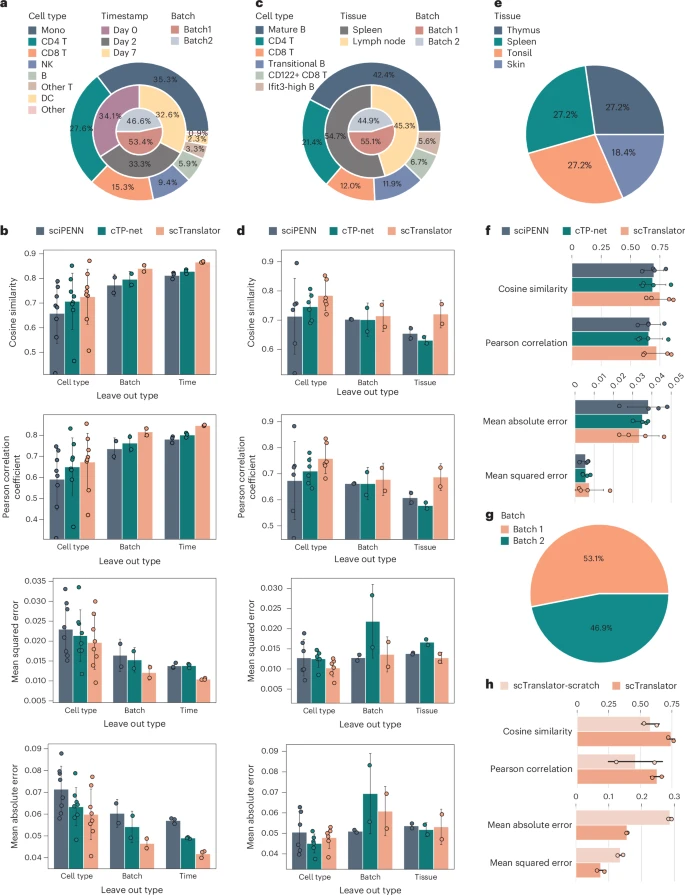
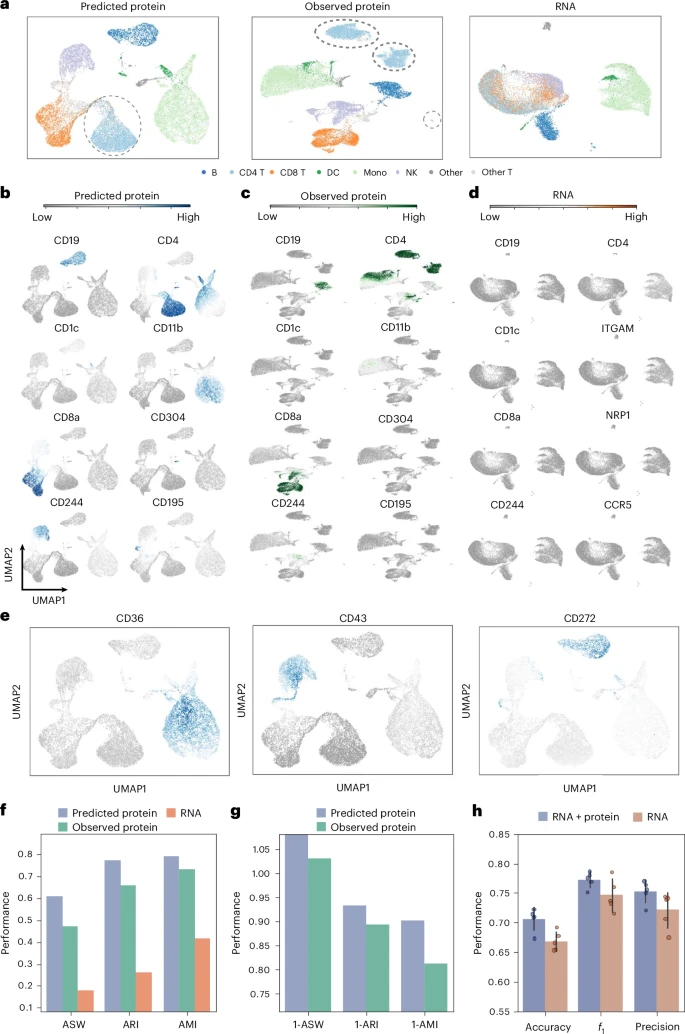
5. 优化下游应用:批次效应校正与细胞分群
伪蛋白质(Pseudo-protein)量化还有助于下游分析。研究发现,由scTranslator预测出的蛋白质丰度在细胞类型识别上表现出更强的对比度和更高的簇内均匀性,比原始的RNA数据更接近真实的蛋白质数据 。
在细胞分群和批次效应(Batch Effect)校正的定量评估中,scTranslator生成的伪蛋白质数据在保持细胞异质性的同时,展现出卓越的批次校正性能,在所有指标上均优于直接观测到的蛋白质数据 。甚至在零样本(Zero-shot)情况下,将预测的蛋白质与RNA数据结合(RNA+protein),相比仅使用RNA数据,在识别肿瘤或正常组织来源细胞的任务中,准确度、F1分数和精确度均有所提升 。
💡 应用展望、局限性与未来路线图:从实验室到临床的加速通道
scTranslator的成功,为单细胞多组学研究打开了一扇新的大门。
潜在的应用场景
完成缺失数据: 鉴于目前单细胞转录组数据海量且易于获取,scTranslator可用于补全缺失的蛋白质组信息,将纯转录组数据集升级为更有洞察力的多组学数据集 。
高通量药物靶点发现: 利用模型的扰动预测能力,研究人员可以在正式的湿实验之前,快速且经济地筛选和验证感兴趣基因对蛋白质水平的潜在影响,从而大幅缩短药物靶点和功能基因的发现周期 。
基因调控网络和蛋白质互作分析: 模型内置的注意力机制可以被用来深入解析基因-基因、基因-蛋白质和蛋白质-蛋白质之间的相互作用关系,帮助我们以更高分辨率的视角理解细胞调控网络 。
局限性与挑战
尽管scTranslator表现出色,但仍存在客观局限性:
零样本学习的偏差: 零样本预测高度依赖预训练数据的多样性和全面性 。如果测试数据包含训练集中未曾捕获到的独特生物学细微差别,预测精度可能会受到影响,存在潜在偏差 。
调控机制的实验验证: 虽然模型通过注意力机制识别了新的调控关系,但这些新发现的调控网络仍需要湿实验室实验进行确认,以验证其生物学上的真实性 。
未来路线图
未来的研究将围绕以下方向展开:
开放资源共享: 研究团队已将模型发现的调控基因列表作为开放资源向生物学界公开,鼓励进行后续的实验验证 。
转录组嵌入工具: scTranslator的编码器经过大量转录组数据的预训练,可以作为一个强大的转录组嵌入工具,为其他下游单细胞分析提供高维、有意义的特征表示 。
泛疾病应用: 随着模型对各种疾病背景下数据的学习和泛化能力的不断增强,scTranslator有望成为阐明癌症等复杂疾病中细胞间关系的有力工具 。
scTranslator无疑是单细胞多组学领域的一个重要里程碑,它以人工智能的力量,正在将曾经耗时费力的蛋白质组测量转化为高效、可计算的“翻译”任务。这一突破,预示着高分辨率、低成本、高通量的细胞世界解析时代,已加速到来。