【诊疗探针】化疗中治疗诊断荧光探针
Theranostic Fluorescent Probes
诊疗一体化荧光探针
化疗的概念起源于1947年首次报道芥子气对淋巴组织具有损伤作用的研究,后续在动物模型(小鼠)中通过氮芥对淋巴瘤的有效抑制得到验证。化疗药物通过抑制癌细胞的增殖速度和缓解肿瘤负担发挥作用,利用癌细胞相较于正常细胞更快的生长速率和增殖特性,实现对前者更显著的抑制作用。随时间推移,多种抗癌药物应运而生,它们通过多样化的机制干预肿瘤发展,有的干扰细胞新陈代谢,有的则针对关键酶进行靶向。文献中记载的大部分药物作用于DNA损伤修复、DNA复制、免疫响应调节及细胞凋亡等关键细胞过程(图3),这些作用机制赋予了药物高度活性,但同时也限制了其对肿瘤组织的特异性,导致非目标组织的毒性副作用,即脱靶效应。为克服这一限制,通过结构改造,产生了治疗探针,这些探针能选择性靶向癌细胞或肿瘤微环境(TME),并在适当条件下释放活性药物(药物本体),即便药物本体本身不具备选择性。掩蔽/去掩蔽策略是实现这一目标的关键,确保了治疗活性仅在癌细胞内释放。类似策略亦应用于可激活的诊疗性荧光探针的开发中。
图3.临床实践中不同类别的化疗药物。G0-休息阶段;G1-生长;S——DNA合成和复制;G2-生长和有丝分裂准备;M-有丝分裂。
肿瘤微环境(TME)与正常组织微环境在多个生理参数上存在显著差异,包括酸性pH值、活性氧(ROS)水平升高、细胞内谷胱甘肽(GSH)浓度增加、特定酶的过度表达以及还原性或乏氧条件,这些特征为区分恶性肿瘤与周围正常组织提供了依据(图4)。
图4.正常组织和肿瘤组织不同微环境示意图[pH=细胞内pH;pHe=细胞外pH,ROS=活性氧]。
本文后续将依据治疗探针在肿瘤微环境中的激活机制,对应用于化疗、光动力治疗(PDT)、光热治疗(PTT)、声动力治疗(SDT)、免疫疗法等多种治疗模式的探针进行细分讨论,以此突出不同策略在精准激活和治疗效果上的独特性。
【化疗探针】pH响应荧光探针
Theranostic Fluorescent Probes
诊疗一体化荧光探针
正常生理条件下的组织pH值维持在大约7.4左右,而肿瘤微环境(TME)的pH值则常常偏低,通常减少0.5至1.0个pH单位,这主要归咎于肿瘤细胞增强的糖酵解作用及在多数实体瘤中常见的缺氧环境,促进了葡萄糖的快速摄取及乳酸的大量产生。肿瘤组织的酸性pH环境在肿瘤的发生、复发、转移扩散及耐药机制中扮演着核心角色。尽管如此,并非所有肿瘤均表现出酸性TME,例如,软组织肉瘤和腺癌的外周微环境pH值(pHe)约为6.94±0.08,而恶性黑色素瘤和鳞状细胞癌的pHe则为7.20±0.07。不管肿瘤类型或阶段如何,pH值的异常通常被视为实体瘤的一种普遍特征,因此,pH值被广泛利用作为癌症靶向递药系统(DDS)的内在触发因子。至今,已有多篇文献报道了在MCF-7、HeLa、和BxPC-3癌症模型中应用pH响应型DDS的研究。
DDS的开发旨在增强药物本身或细胞毒性剂的治疗效果,同时最大限度地减少不良副作用。实现这些目标要求DDS既要有足够的稳定性以确保药物的运输和循环,又能在目标位置高效释放药物。在基于pH触发的DDS中,溶酶体、内体、细胞或肿瘤组织局部pH值的下降是启动药物释放的关键因素。近年来,众多研究报告了利用这种酸性pH响应机制的治疗系统。这些系统通常将药物通过酸敏感的化学键如乙醛、肼、肟或亚胺相连。这些化学键在生理pH(7.4)下稳定性良好,但在酸性内体环境中则快速水解,故能特异性地在酸性肿瘤部位释放药物。然而,这种策略主要适用于含有可利用的游离醛基或酮基的药物,限制了其适用范围。
基于腙/肟/亚胺的治疗诊断探针
酸催化水解机制,尤其是在腙和肟连接体的应用上,已经成为构建pH敏感型药物递送系统(DDS)的基石技术,尤其在输送小分子药物、设计纳米载体与聚合物基递药体系以精准导向肿瘤部位方面展现出巨大潜力。Kalia等人通过详尽的氚标记缓冲液核磁共振研究揭示,亚胺碳上的亲核水分子进攻是决定药物分子从腙或肟结构水解脱速率的关键步骤。此过程伴随亚胺氮的质子化,最终促使药物分子自由释放(见图5)。吸电子取代效应能抑制氮原子的质子化倾向,从而调控整体水解速率。在生理pH7.4条件下,连接体的稳定性序列为三烷基肼显著高于>肟>酰基腙>伯腙>仲腙>亚胺。鉴于季铵盐型肼在pH5时表现出的高度稳定性,其不太适合酸敏感递药应用。相反,酰基腙连接体在中性pH环境下高度稳定,且在酸性条件(pH5)下不易降解,因此成为极具吸引力的选择。
图5.基于腙的pH响应治疗诊断剂及其应用
阿霉素(Dox),作为一种蒽环类药物,广泛应用于包括乳腺癌、肺癌、卵巢癌等多种癌症以及多种儿童血液肿瘤的治疗中。但其临床应用受限于诸如疲劳、肝脏和心脏毒性等副作用。为克服这些局限,研究者们探索了多种策略,其中改进Dox至肿瘤部位的递送方法是关键。例如,通过酸敏感的腙连接体将Dox与靶向基团的GRDS-寡肽及香豆素分子偶联,形成诊疗试剂1(见图5)。Dox本身具有595nm(激发于470nm)的固有荧光特性,这使其在癌症成像领域得到广泛应用。在复合物1中,Dox的荧光因与香豆素分子的相互作用而被淬灭,直至pH5条件下释放Dox。在整合素阳性的U87胶质母细胞瘤细胞中,观察到剂量依赖性毒性(IC50=0.19μgmL-1),同时释放Dox前后的荧光变化(Dox的红色荧光和香豆素的蓝色荧光)可实时监测药物定位与激活。
在骨架设计中整合细胞凋亡标记物,能够无创评估细胞毒素的激活与定位,从而实现诊疗试剂量的精细化调整。诊疗试剂2利用双FRET机制,展示了这一策略(见图5)。该诊疗试剂通过酸不稳定腙键将抗癌药物Dox与强荧光淬灭剂Dabcyl和Caspase-3响应肽序列(DEVD)结合,同时引入了FAM荧光团,以便在细胞层面实时监测药物活化。该复合物还携带靶向特异性RGD序列,以增强癌症靶向性。体外研究表明,诊疗试剂2在pH5环境下释放超过90%的活性Dox,而在生理pH7.4下释放率显著降低(仅19%)。在U87细胞中,2号诊疗试剂随时间呈现荧光增强,归因于DEVD肽被裂解后FAM荧光的释放,伴随明显的治疗效果(IC50=4.3×10-6M)及Caspase-3激活。
为简化结构同时保持癌症特异性激活与药物释放的实时监测,研发团队开发了诊疗试剂3(图5),通过腙键连接Dox与荧光团BODIPY。该复合体在生理pH下荧光较弱,而在酸性环境(pH6.5-4.5)中,腙键断裂释放出游离BODIPY,引发显著荧光。以RAW264.7巨噬细胞为模型,通过脂多糖(LPS)处理诱导促炎性M1巨噬细胞,导致酸化,3号诊疗试剂处理显示剂量依赖性毒性及荧光“开关”效应。在IL-4介导的抗炎M2巨噬细胞中未见此现象。类似效应在LPS处理的斑马鱼模型中亦得到验证,凋亡的巨噬细胞展示出与Dox对应的红色荧光,邻近区域则发出BODIPY对应的绿色荧光。诊疗试剂3还在斑马鱼体内再生模型中测试,以追踪吞噬性M1巨噬细胞的活性,与LPS联用能促进M1极化,有利于组织修复。
诊疗试剂4的设计旨在提升Dox在癌细胞中特异性摄取的能力,并与正常细胞相比进行了测试(图6)。Kim等人利用Dox的固有荧光监控前药活化与细胞内分布,采用硝基苯分子通过腙键连接Dox作为PET荧光淬灭剂,并嵌入生物素单元实现癌症靶向。在酸性环境下,腙键水解引起荧光“开启”,释放自由Dox的荧光信号。体外实验显示,4号诊疗试剂在生物素阳性的HepG2癌细胞中相较于生物素阴性的正常WI38细胞显示出高度选择性。
图6.酸敏感治疗诊断剂的化学结构及其应用
同一研究团队进一步研发了一种复式策略,旨在运用双重成像模态——荧光成像与磁共振成像(MRI)——来同步评估细胞的摄取效率与激活动态。这一创新策略通过化合物5(图6)得以展现,其中两个荧光标记的阿霉素(Dox)单元与一种顺磁性钆配合物——水溶性texaphyrin莫替沙芬钆(MGd)——经由腙键桥接。在酸性环境下,复合物5的荧光强度减退,这一现象在癌细胞株A549与CT26中亦得到确认,而正常成纤维细胞NIH3T3即便在高达100μM的浓度下也未显现显著毒性。化合物5在磷酸盐缓冲盐水(PBS)中的T1对比弛豫率分别达到20.1±0.4mM(-1)s(-1)(60MHz)和6.1±0.2mM(-1)s(-1)(200MHz),显著超越了常规Gd3+造影剂。此外,在仅4μM的低浓度下,经5处理的细胞模型便在T1弛豫时间上迅速饱和,凸显了其高效性(见图6)。另一款针对肿瘤的诊疗试剂6,则通过腙键将靶向序列AP2H(IHGHHIISVG)与Dox相连,设计上确保在弱酸性条件下降解,对LAPTM4B阳性的肺癌细胞A549(IC50=1.14μM)和肝癌细胞HepG2(IC50=4.0μM)呈现出剂量依赖性毒性。荧光分析显示,6在这些细胞内首先于内体和溶酶体中激活,继而转移至细胞核(图6),而在非癌性的HEK293细胞中激活程度极低。
尽管基于肼的连接方式在小分子系统中应用广泛,基于肟的连接方式的研究虽少,却在聚合物体系中逐渐崭露头角。本文侧重于基于单体分子的治疗体系。Jin等人利用对苯二甲醛在PEG-Dox胶束中构建肟连接方式,观察到pH5.0时水解速率缓滞(t1/2=15小时),而中性pH条件下药物释放效率更高(12小时内释放20%),提示肟连接方式的稳定性可能低于酰基腙类。Xu等则设计了一种右旋糖酐-Dox共轭物7(图7),旨在针对人肝癌HepG2细胞进行酸敏感的Dox递送。在pH7.4的中性环境下,72小时内约25.9%的Dox释放;而在更低pH值下,肟水解加速,导致Dox释放显著增加(pH6.8时为40.4%,pH6.0时达64.7%,pH5.0时高达87%)。通过激光共聚焦显微镜和细胞活力检测,确认了7在HepG2细胞中的内吞摄取及其在酸性条件下的进一步水解。7处理后的细胞毒性(IC50=0.73μgmL-1)与游离Dox(IC50=0.62μgmL-1)相近。在H22异种移植小鼠模型中,7的静脉注射治疗显著抑制了肿瘤生长(抑制率71.0%),提高了生存率,并减轻了副作用,优于其他对照组,包括游离Dox治疗(抑制率45.4%)和7的还原类似物(抑制率19.2%)。
图7.酸敏感治疗诊断剂的化学结构及其应用
其他酸响应治疗诊断探针
Yang等人巧妙地设计了一种名为N-乙氧基苄基咪唑(NEBI)的酸敏感调节性化学连接方式,用于叶酸受体阳性癌细胞中茚异喹啉类抗癌药物的特异性递送。该连接方式的核心是基于酸催化的“氨基醇”醚官能团水解,其中结构8利用咪唑环氮原子构建了这一关键结构(图7)。在酸性微环境诱导下,咪唑环的质子化促进了药物分子的自主释放。通过调整苯环上的取代基,可以调控水解速率和药物释放速度:引入吸电子基团(如硝基)会显著降低水解速率(t1/2=6900小时,pH5.5),而富电子基团(如甲氧基)则加速药物释放(t1/2=0.6小时,pH5.5)。重要的是,无论取代基类型如何,药物本身在生理pH值(7.4)下的稳定性均显著高于酸性条件(pH5.5)。体外研究表明,针对叶酸阳性的KB细胞,该策略实现了药物的有效靶向递送,其IC50值为60μM,而去除叶酸受体则导致药效大幅降低(IC50=655μM)。尽管PEG修饰降低了药物在KB细胞中的活性(IC50=250μM),但由于药物自身的荧光特性,结构8能够通过荧光显微镜直接追踪其细胞内摄取和活化过程,彰显了NEBI连接方式通过叶酸受体介导内吞途径递送药物的巨大潜力。
此外,马来酸衍生物作为一种酸敏感连接方式,在药物递送领域也展现出应用价值,其释药机制依赖于在pH低于羧酸pKa时发生的内环化反应。早先研究聚焦于顺式乌头酸酐的构建,但产物常伴随脱羧和几何异构化的问题。以Dox为例,前药9通过马来酰胺酸连接(图7),在酸性环境下形成中间体10,该中间体在pH6.0时能释放约70%的Dox。Dox分子本身的游离氨基(pKa=8.2)对酸性肿瘤环境极为敏感,导致其在pH6.7时对ES-2卵巢癌细胞的毒性(IC50=1.6±0.1μM)高于中性pH7.4(IC50=0.7±0.1μM)。而基于马来酰胺酸的连接方式10在不同pH值下的效力较为恒定(pH6.7时IC50=2.1±0.3μM,pH7.4时IC50=3.2±0.4μM),尽管其活性略低于Dox,却有效缓解了Dox的毒性。Dox的天然荧光特性同样使化合物11成为一种便于监测药物活化与定位的酸敏感诊疗试剂。
综上所述,酸敏感化学连接方式持续推动着基于小分子的递药系统的发展,特别是在肿瘤微环境(pH6.0-7.0)中,酸催化的连接方式能够有效释放药物。针对不同递送目标,如实体瘤或溶酶体、内体等酸性亚细胞器(pH4.5-6.0),选择合适的酸敏连接方式至关重要,如马来酰胺酸类连接方式对微小pH变化的敏感性使其在实体瘤递送中更具吸引力,而酰基腙类连接方式则更适合酸性更强的溶酶体环境。显然,对化学连接方式的进一步研究与优化将是提升癌症靶向治疗递送效率的重要途径。
【化疗探针】GSH响应荧光探针
Theranostic Fluorescent Probes
诊疗一体化荧光探针
在复杂的生物环境中,多种氧化还原过程在细胞内外及不同组织中频繁上演,其中的典型代表包括NADP+/NADPH、O2/O2--、硫氧还蛋白(TrxSS/Trx(SH)2)和谷胱甘肽(GSH/GSSG)等氧化还原对。谷胱甘肽,因其细胞内高浓度(1-10mM)及其在维护细胞完整性、介导细胞功能、代谢和凋亡中的关键作用,成为治疗和递药领域关注的焦点。GSH作为关键的抗氧化剂,有效阻止ROS介导的损伤,保护细胞免受氧化应激。然而,GSH在细胞外基质、血液及细胞表面的浓度显著偏低(2-20μM),这与该区域富含能稳定二硫键的蛋白质有关。细胞内环境则通过NADPH和GSH还原酶系统维持还原状态,确保GSH主要以还原形式存在。特别值得注意的是,细胞内部存在显著的GSH浓度梯度。有趣的是,肿瘤组织中GSH水平较正常组织高出约4倍,这与癌细胞快速增殖有关,虽然高GSH水平可能削弱多种癌症(如卵巢癌、乳腺癌等)的治疗效果,但这也为设计基于氧化还原敏感的药物递送系统提供了契机,尤其是在肿瘤微环境中特异性释放活性药物或荧光探针。
基于GSH的还原性,二硫(S-S)键是最常用的化学连接方式之一,能够在存在游离硫醇的情况下通过硫醇-二硫交换反应断裂。GSH因此成为激活含二硫键前药的理想生物催化剂。在前药设计中,采用抗体、肽段、小分子等多种载体,对基于二硫化物的策略进行了广泛探索。特别是在小分子诊疗学中,众多候选药物和荧光团,如Dox、喜树碱、紫杉醇、吉西他滨、萘啶酰亚胺、hemicyanine、dicyano-methylene-4H-pyran、荧光素等,已通过三种主要策略与GSH响应性二硫键相连:(i)利用可裂解的连接方式连接化疗药物与荧光团;(ii)连接具有固有荧光的化疗药物与靶向配体;(iii)通过多组分策略结合化疗药物、荧光团和靶向配体(图8)。
图8.GSH响应可激活分子治疗诊断探针的示意图。
以(i)类策略为例,诊疗试剂12通过S-S连接喜树碱(CPT)与半氰基荧光团,用于监测H22肿瘤小鼠模型中GSH介导的激活过程,其中CPT的初始荧光被抑制,但在GSH作用下,二硫键裂解,释放CPT并伴随着荧光信号的增强(图9),类似的策略被广泛用于监测不同肿瘤模型中的GSH激活情况。
图9.GSH激活治疗诊断探针的效果。
对于(ii)类策略,具有固有荧光的抗癌药物通过二硫键与癌症靶向配体相连,如叶酸-阿霉素诊疗试剂13,其设计中α,α-二甲基取代的对噻吩基脲烷二硫化物增强了稳定性,且在模拟癌细胞质的GSH浓度下表现出良好的药物释放动力学(图8,9)。
(iii)类策略通过添加癌症靶向单元提升药物的特异性并降低全身毒性,如RGD肽修饰的萘二甲酰亚胺原喜树碱诊疗试剂14,通过RGD与U87癌细胞的avβ3受体结合,实现GSH触发的药物释放与荧光成像(图8,9)。此外,还有如诊疗试剂15和16等,分别用于吉西他滨的GSH选择性递送和Cy7-吉西他滨的叶酸靶向递送,均显示了通过GSH敏感连接方式在特定肿瘤细胞中激活的潜力和细胞内定位的可视化(图8,9)。
这些策略也拓展到了肽类抗癌药物的靶向递送,如17,通过二硫键连接策略,实现了对HepG2细胞的选择性递送和荧光信号增强,证明了基于肽的诊疗试剂在改善递送、增强治疗靶向性和成像跟踪方面的潜力(图10)。综上,这些研究不仅加深了对GSH在氧化还原敏感性递送系统中作用的理解,也为开发更高效、更精准的癌症治疗策略提供了坚实的基础。
图10.GSH响应治疗诊断探针17-22的化学结构和效果
除二硫化合物外,研究者亦涉足其他化学键在GSH响应性递药领域的应用,其中硒元素因与硫同族且化学性质相近而备受瞩目。硒的氧化还原特性尤为突出,其Se-Se键的断裂能(172kJmol-1)远低于C-Se键(244kJmol-1)或S-S键(268kJmol-1),这使得硒-硒连接方式相较于含硫类似物展现出更高的GSH敏感性。例如,Fang等人设计的两种硒化喜树碱衍生物诊疗试剂18和19(图10),通过HPLC分析确认了它们在GSH存在下于430nm处(激发波长365nm)增强的荧光释放特性。实验揭示,在1m MGSH的Tris-EDTA缓冲液中,两者的药物释放几乎瞬时完成。释放出的硒酚中间体预期可通过消耗细胞内GSH并促进ROS生成,增强18和19的治疗效果。动物实验中,HepG2异种移植瘤模型在接受18或19治疗后,肿瘤重量减少约75%,超越了标准CPT治疗效果的53%。尽管成果显著,基于硒的连接方式因合成难度及对GSH与ROS的双重敏感性可能引起的非预期活化和药物泄露问题,目前使用并不普遍。
此外,生物硫醇如半胱氨酸(Cys)和同型半胱氨酸(Hcy)在调控细胞氧化还原稳态中同样扮演关键角色。Cys的缺乏与多种疾病相关,涵盖心血管疾病至造血功能障碍等。鉴于某些GSH敏感连接方式对Cys亦有响应,这在Cys和GSH共存的生理环境下可能导致非目标区域的药物误激活。反之,专一针对Cys的递药系统则可实现治疗的精准性。近期,2,4-二硝基苯磺酰(DBS)作为Cys敏感连接方式在递药领域崭露头角,尤其适合与含有羟基或胺基的药物分子结合。Jo和Johansson等团队分别利用DBS直接修饰伊立替康活性代谢产物SN-38(诊疗试剂20)和阿霉素(DOX,诊疗试剂21)(图10),并观察到DBS引入后药物的荧光被显著淬灭。
在还原性环境下,DBS经历芳烃亲核取代反应,释放SO2,同时激活药物并增强荧光。Wu等人研发的诊疗试剂22b,巧妙结合了硫醇响应性DBS触发机制、喜树碱(CPT)及近红外荧光团二氰亚甲基-4H-色烯(DCM),展示了增强的多功能性。该复合物22b在未活化状态下无活性,但在硫醇触发下,通过级联反应释放CPT并恢复DCM的近红外荧光。诊疗试剂22b在HeLa癌细胞(IC50=5.8μM)和L929细胞(IC50=8.9μM)中展现出增强的剂量依赖性细胞毒性,优于无药物对照22a。22b的脂质体诊疗试剂在体内给药后,有效抑制了肿瘤生长,进一步证实了其治疗潜力。
【化疗探针】硫化氢响应治疗诊断探针
Theranostic Fluorescent Probes
诊疗一体化荧光探针
硫化氢(H₂S),以其易燃性、标志性臭鸡蛋气味及腐蚀特性而著称,是一种具有潜在毒性的气体。类似于一氧化碳(CO)和一氧化氮(NO),内源性H₂S作为气体信号分子,在调控生理活动如细胞分化、增殖、存活/凋亡及代谢过程中扮演关键角色。H₂S的代谢失衡与多种疾病状态紧密相关,涵盖了癌症、阿尔茨海默病及糖尿病等重大健康挑战。H₂S可以通过提升谷胱甘肽(GSH)水平以对抗氧化应激,间接参与清除自由基(ROS),保护神经元免受损害。这一独特属性激励科学家们探索H₂S响应型药物的设计,视其为癌症治疗的新型策略。值得注意的是,H₂S不仅展示出明显的亲核性和还原性——特别是在其脱质子形态下——还能特异性催化叠氮化物转换为胺类化合物,此胺类的高度电子富集状态可触发荧光团或药物的有效释放。
在此背景下,Qian等人设计了一种新的H₂S响应性诊疗试剂23(图11),该诊疗试剂整合了抗肿瘤药物氨酰胺,用于胶质母细胞瘤的精确治疗。氨酰胺的自然荧光因PET效应而被抑制,然而在遇到H₂S后,其携带的叠氮基团经还原转化为胺,随后通过1,6-消除机制经由硫醇连接方式断裂释放出活性药物。荧光成像技术表明了该药物在进入细胞内后首先进入了溶酶体,并进一步转移至细胞核,通过诱导DNA损伤和线粒体功能障碍机制展现细胞毒性。在U87MG三维细胞球模型中,该诊疗试剂即便在低剂量(30μM,处理两天)下也能表现出显著疗效,其破坏肿瘤球体完整性的能力优于传统化疗药物顺铂(40μM,处理两天)。相似的策略还应用于构建探针24(图11)。
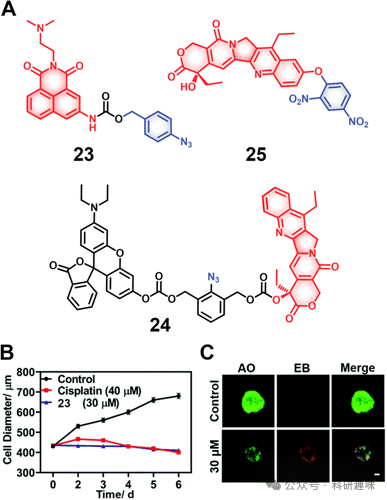
图11.(A)硫化氢响应治疗诊断探针23-25的化学结构及其效果
鉴于硫化氢(H₂S)的固有亲核性,Li及其研究团队创新性地设计了诊疗试剂25(图11),该分子巧妙地将抗癌活性成分SN-38与一个强吸电子基团——二硝基苯(DNP)通过醚基团链接,形成了一个智能型前药结构。在未触发状态下,诊疗试剂25因分子内电荷转移(ICT)效应被抑制而不具备活性。然而,在H₂S的环境下,稳定的醚键断裂,伴随这一过程的是荧光信号显著增强,同时SN-38得以释放,恢复其生物活性。实验研究证明,诊疗试剂25能在H₂S浓度升高的HCT116与4T1肿瘤细胞系中实现选择性摄取与激活,展现出高效细胞杀伤效果。尤为值得一提的是,SN-38固有的荧光特性使药物的动态激活过程得以实时可视化监控,为疗效评估提供了直接证据。
该策略巧妙利用H₂S的亲核性作为激活开关,为前药活化领域提供了一个新的模式。未来研究需深入探讨在多种亲核物质共存条件下,如何保持H₂S激活路径的高度选择性,这是优化此类设计的关键。尽管目前文献报道显示该方向前景乐观,但仍需注意,基于H₂S亲核性激活机制的治疗药物开发尚属新兴领域,其潜力和局限性有待进一步探索和明确。
【化疗探针】过氧化氢响应治疗诊断探针
Theranostic Fluorescent Probes
诊疗一体化荧光探针
"ROS"这一术语指的是由分子氧(O2)还原形成的相对不稳定的分子和自由基集合,主要包括过氧化氢(H2O2)、超氧阴离子(O2-)、单线态氧(¹O₂)和羟基自由基(·OH)。细胞内ROS主要源自线粒体呼吸链活动和NADPH氧化酶催化的反应过程。此外,外部因素如紫外线照射和异生物质也可诱导ROS的生成。适量ROS对于维持细胞稳态至关重要,它们作为信号分子参与调控细胞生长、增殖、迁移及凋亡等基本生命活动。ROS通过调节特定蛋白质活性,还与血管功能、氧感应、免疫反应和基因表达调控等生理过程紧密相关。然而,ROS过量可引发氧化应激,导致核酸、脂质及蛋白质损伤,与多种疾病如衰老、癌症、心血管疾病、糖尿病及神经退行性疾病的发生发展密切相关。
线粒体中,H2O2由超氧阴离子通过超氧化物歧化酶(SOD)转化生成,其在炎症细胞和癌细胞中的产生速率(约0.5nmol/104cells/h)显著高于正常细胞(约0.050±0.004nmol/104cells/h)。作为其他高活性ROS前体,H2O2以其较长的半衰期(t1/2=1ms)和在氧化应激情境下累积的特性,成为了ROS响应型递药系统中备受瞩目的激活因子,尤其考虑到大多数ROS半衰期极短(t1/2<1μs)。在设计癌症特异性探针方面,多种化学基团如芳基硼酸酯、硫醚/硫酮、氨基丙烯酸酯、硒/碲化合物及聚脯氨酸等,已被探索用于响应H2O2。本节综述了在氧化应激(特别是H2O2触发下)激活的治疗探针进展。
Kim等人开发的探针26(图12)是一个双药单元系统,其中5′-脱氧-5-氟尿苷(5-FU的前药)通过双苯基硼酸与乙脒荧光基团(作为线粒体凋亡指示剂)相连。由于乙脒部分带正电,诊疗试剂26在A549肺癌细胞中优先内化并富集于线粒体中。肿瘤细胞内较高的H2O2水平促使5′-脱氧-5-氟尿苷释放,进一步被胸苷磷酸化酶转化为活性药物5-FU。伴随乙脒与DNA的插入,释放出的自由乙脒和荧光增强共同作为荧光报告信号,实现实时监测细胞凋亡过程。在A549异种移植小鼠模型中,26通过尾静脉注射后,其在肿瘤组织中的荧光信号增强,与H2O2介导的探针激活相符。注射脂多糖(LPS)进一步增强荧光输出,进一步验证了氧化应激触发的机制。与对照组(PBS处理)和其他实验组(5′-FU直接处理)相比,治疗探针26显示了肿瘤特异性聚集和显著的肿瘤抑制效果。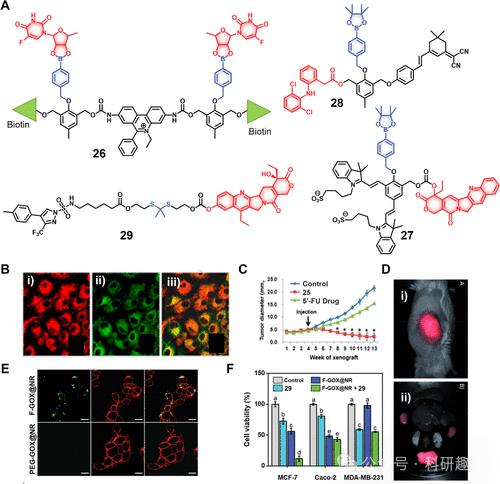 图12.(A)H2O2响应性治疗诊断探针的化学结构(26–29)与效果
图12.(A)H2O2响应性治疗诊断探针的化学结构(26–29)与效果
Shabat及其团队运用了一种近红外荧光探针Cy7,以实现药物活化与定位的精准监测。他们构建了诊疗试剂27(见图12),该分子由硼酸酯基团与氰基荧光基团及喜树碱(CPT)共价结合而成。在H2O2环境下,硼酸酯结构发生断裂,继而引发一连串反应,在约90分钟内逐步释放出游离的活性CPT与Cy7,同时在720nm波长处出现增强的荧光信号。实验结果显示,在U87细胞系中,27在H2O2作用下表现出较高的细胞毒性(IC50=40nM),而在无H2O2条件下则毒性大减(IC50=250nM)。
尽管27相对于药物本身CPT(IC50=20nM)的直接毒性来说有所降低,但因其减少了非特异性毒副作用,故被视为个性化医疗策略中的潜在策略。另一个与之相近的近红外探针28被设计用于H2O2介导的双氯芬酸(一种非甾体抗炎药物)在炎症环境下的巨噬细胞释放与监测。尽管苯基硼酸酯作为H2O2响应链接子被广泛应用,其在酸性条件下的不稳定性却可能限制了应用范围,导致药物在溶酶体等酸性细胞器中的非特异性激活。为解决此稳定性难题,YanLi和Nam团队创新性地引入了硫酮基链接体,该链接体在不同pH环境下均表现出增强的稳定性,其氧化裂解产物——硫醇和酮,成为药物递送体系研究的焦点。
鉴于H2O2主要在线粒体中生成,治疗药物若无法有效聚集于此,则可能减弱抗癌效果。Landfester等人的研究提出了一种新颖策略:直接在癌细胞线粒体中原位生成H2O2。他们设计了复合型治疗探针29(图12),由塞来昔布修饰的SN-38通过H2O2敏感的硫酮连接方式相连,并封装于叶酸修饰的二氧化硅纳米颗粒内,该颗粒表面固定有葡萄糖氧化酶(GOX),形成了所谓的纳米反应器F-GOX@NR。此纳米反应器经叶酸受体介导的内吞作用进入癌细胞后,GOX能催化葡萄糖转化为H2O2,进而诱导塞来昔布-SN-38共轭体中硫酮键断裂,释放出活性SN-38。随后,SN-38作为抑制剂引发细胞凋亡。对比实验揭示,F-GOX@NR+29nm反应器体系在叶酸和COX-2双阳性的MCF-7细胞中表现出显著增强的协同治疗效果(IC50=0.14μM,24小时),远优于单独使用SN-38(IC50=1.2μM,24小时)或塞来昔布-SN-38偶联物(IC50=2.8μM,24小时)。此外,SN-38固有的荧光变化(在H2O2作用下,最大发射峰由452nm移至560nm)提供了实时监测药物活化与分布的便利途径。
【化疗探针】其他ROS响应性治疗诊断探针
Theranostic Fluorescent Probes
诊疗一体化荧光探针
除过氧化氢(H2O2)之外,其他活性氧物种(ROS),包括单线态氧(¹O₂)和羟基自由基(-OH),因其高度反应性,亦被视为潜在的药物释放激活因子。-OH以其广泛的反应性著称,能与氨基酸、碳水化合物、脂质及核酸等多种生物分子无选择性地发生作用。相比之下,¹O₂作为氧气的激发状态,氧化能力更为强劲。在生物体系内,-OH通常经由过渡金属离子(如Cu(I)或Fe(II))催化的H2O2分解过程,即芬顿反应产生,而¹O₂主要由光敏剂(PSs)生成,详情参见后续2.2节。值得注意的是,¹O₂与-OH在生理环境中的半衰期均较H2O2短暂。
Zhang及其团队设计了一种治疗探针30(图13),通过ROS响应性的硫代金属连接方式,将吉西他滨与红光激活的荧光光敏剂(间-四苯基卟啉,TPP)相结合,旨在实现荧光成像引导的治疗策略。此探针中,药物的5′-OH末端经修饰以暂时抑制其抗癌活性。鉴于¹O₂的超短半衰期(≈40纳秒)及有限的扩散距离(20-200nm),在非光照条件下,细胞内微量的¹O₂不足以引发明显的细胞毒性。然而,在658nm光照射(280mW/cm)下,TPP作为光敏剂生成¹O₂,触发硫代金属键断裂,继而释放药物。释放的药物不仅在光照区域自由扩散,还在邻近未直接光照的细胞中引起毒性。诊疗试剂30在光照下对HeLa细胞展现出了浓度依赖的毒性(IC50=0.25μM),优于对照组(TPP、TPP-UCL-GEM)。在H22小鼠肿瘤模型中,PEG2000-PLA2000修饰的30经静脉注射后,能富集于肿瘤并经低功率光照抑制肿瘤生长,且利用TPP的自然荧光追踪了体内分布情况。相似地,Wang等人在乳腺癌小鼠模型中实现了Dox的有效递送,且副作用微乎其微。
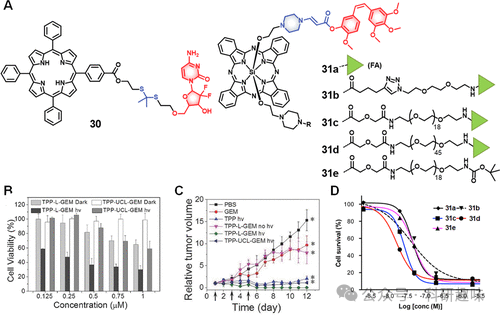
图13.(A)ROS响应性治疗诊断探针的化学结构(30和31)及其效果
这方面的研究还涉及利用¹O₂反应激活其他治疗药物,如采用氨基丙烯酸酯类连接方式桥接光敏剂与药物本身。这一策略便于含羟基药物通过羧酸与氨基丙烯酸酯偶联,或通过氨基丙烯酸酯的氨基端与PS相连,构建多样化治疗载体。据此方法,研发团队已开发出多种装载SN-38、康维司汀、紫杉醇、以及非甾体抗炎药(如布洛芬和萘普生)的递送系统。
You等人开发的一系列诊疗试剂31a-e(图13),这些共轭物包含远红外激活的光敏剂(硅酞菁,Pc),通过氨基丙烯酸酯与紫杉醇在2′-OH关键位置共价结合,该位置涉及与微管蛋白的相互作用,对于药物功能至关重要。这些共轭物进一步与不同分子量的聚乙二醇(PEG,1kDa至5kDa)和叶酸偶联,以增强靶向性和溶解度。研究确认,中等长度的PEG链(1k至3.5kDa)最适于促进叶酸受体介导的摄取。实验显示,与长链PEG或无PEG修饰的共轭物相比,这类中链PEG化共轭物展现出更强的细胞毒性。在Colon-26细胞的实验中,共轭物31b(携带2kDaPEG)在690nm光照下展现出最高的细胞毒性(IC50=1.65nM),优于其他共轭物(IC50=2.71至4.85nM)(图13)。
【化疗探针】酶响应治疗诊断探针
Theranostic Fluorescent Probes
诊疗一体化荧光探针
酶,作为生物催化剂的杰出代表,主要是由复杂蛋白质结构构成的大分子,它们在细胞内外催化各类化学反应,极大地促进了生命过程的速率。这些生物酶凭借其卓越的底物专一性、高效的催化活力以及对特定细胞环境的适应性,展现了作为精细化学反应调控工具的巨大潜力。事实上,关键酶活性的异常调节已成为诸如炎症、癌症及神经退行性疾病等多种疾病发病机制的核心环节。
鉴于此,通过精巧设计酶的特异识别底物,科研人员能够巧妙地封装或伪装抗癌药物,从而打造出一类崭新的治疗探针。此类探针不仅能够确保药物仅在目标位点被特异性酶激活,增强了治疗的精准性,还有效提升了药物的体内稳定性,并且在激活后展现出更为显著的治疗效果。以下内容将概述在这一创新领域中所取得的研究进展与成就。
DT-黄酶响应治疗诊断探针
醌是一类在多种天然产物及合成或半合成化合物中频繁出现的核心结构单元,这类化合物包括但不限于抗癌药物、抗菌剂、染料、维生素K以及酶辅因子等。醌类化合物因其还原性特点,在氧化还原循环中扮演着核心角色。醌还原酶1(NQO1),又名DT-二磷酸还原酶,是一种主要分布于细胞质,且在线粒体和内质网中少量存在的双电子转移酶。该酶参与解毒机制,与肿瘤发生的早期阶段紧密相关,在卵巢癌、甲状腺癌、乳腺癌、结直肠癌和胰腺癌等多种癌症类型中发现其表达水平显著升高。正是由于在癌组织中NQO1的高表达,与正常组织形成鲜明对比,使得该酶成为肿瘤特异性药物递送系统中极具吸引力的内源性激活因子。
鉴于吲哚醌作为NQO1的已知底物,Nishimoto等人设计了诊疗试剂32a(图14),旨在利用这一特性向癌细胞输送细胞毒性药物SN-38,并利用DT-二磷酸酶介导的反应实现药物的可控释放,同时伴随荧光信号的变化。为了进一步增强对癌细胞的靶向性,研发了诊疗试剂32b,该分子结合了SN-38与吲哚醌,并通过整合素特异性肽序列作为靶向模块。诊疗试剂32b在表达αvβ3整合素的癌细胞中显示出优先吸收能力,其机制推测为通过NQO1介导的还原反应释放活性SN-38,并在人宫颈癌KB细胞株中产生了50-70%的生长抑制作用。释放的SN-38形成的烯酰亚胺中间体对DNA的烷基化活性被认为是引起细胞毒性的关键机制。
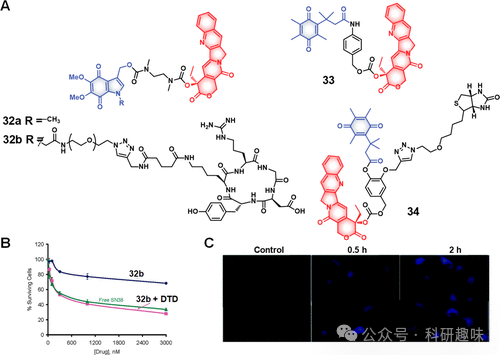
图14.(A)DT-心肌黄酶响应治疗诊断探针的化学结构。(B)治疗诊断探针32b、SN-38和32b+DTD在KB细胞中的细胞活力。(C)在不存在和存在治疗诊断探针33(10μM,2小时)的情况下,在不同时间点记录的A549细胞的荧光图像。
此外,一些研究工作聚焦于基于醌的前药设计,以期优化药物释放特性。Wang等人开发的一种含有“三烷基锁”的醌衍生物尤其引人注目,该结构在DT-二磷酸酶还原作用下转化为对苯二酚,通过内酯化形成稳定的六元环结构,从而促进连接于对苯二酚羰基上的药物有效负载的释放。这一设计理念在Wu等人报道的诊疗试剂33中得到应用,该药物递送系统旨在向癌细胞输送SN-38,且SN-38的内在荧光特性使药物释放和治疗效果的实时监测成为可能(图14)。Kim等人则在诊疗试剂34中引入了额外的功能性标记——生物素,作为癌症靶向基团,该系统展现出对癌细胞的优先摄取能力,并在假设的药物激活后增强了治疗效率。
偶氮还原酶响应治疗诊断探针
偶氮还原酶,作为黄素依赖性酶家族的一员,在真核生物和细菌中普遍存在,多数定位于细胞膜,对于维持生物体内环境的稳态发挥着不可或缺的作用。这些酶通过利用NADH或NADPH作为电子供体,催化特定底物的还原反应。值得注意的是,偶氮还原酶在多种癌症(如肺癌、乳腺癌及胰腺癌)中呈现异常高表达,这一特性启发了科研人员探索以偶氮还原酶为靶点的治疗药物开发。
Kim等人设计的诊疗试剂35(图15)巧妙地利用偶氮连接策略,旨在将化疗药物精确递送至癌细胞线粒体中。该诊疗试剂由罗丹明123类似物与氮芥类似物N,N′-双(2-氯乙基)-1,4-苯二胺通过偶氮键桥接,并融入亲脂性三苯基膦基团以导向线粒体。在偶氮还原酶作用下,偶氮键断裂,同步释放出活性化疗药物与荧光示踪分子,为在乏氧肿瘤微环境中药物活化和定位提供了可视化的手段。尽管如此,35的多步骤合成复杂且溶解性不佳,限制了其转化应用潜力。针对这些问题,Xie等人推出了诊疗试剂36(图15),通过在结构中整合带正电荷的荧光团以确保线粒体靶向性,并减小极性表面积以改善水溶性,从而优化了设计。
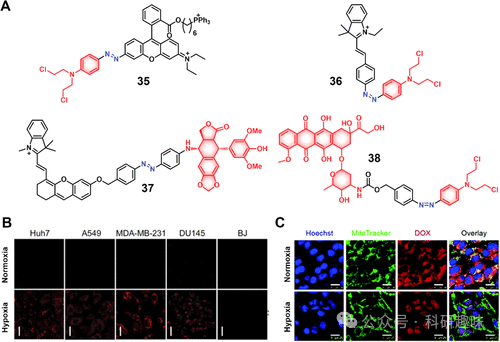
图15.(A)偶氮还原酶响应治疗诊断探针的化学结构(35–38)。(B)不同细胞系中常氧(21%)和乏氧(3%)条件下治疗诊断探针35的荧光图像(比例尺=10μm,激发=555nm,发射=585nm)。(C)使用Hoechst33342和MitoTrackerGreen染色后,在常氧和乏氧条件下用治疗诊断探针38(20μM)处理后记录的4T1癌细胞共聚焦图像(比例尺=20μm)。
沿袭相似思路,Shi团队开发的诊疗试剂37(图15)采用了Cy作为近红外荧光探针,适用于活体药物跟踪。另一方面,Yu等人设计的治疗系统38(图15)将氮芥与阿霉素(Dox)通过偶氮键相连,在还原性条件下,两者得以释放,伴随Dox释放产生的荧光增强,为药物活化及细胞内分布提供了实时监测指标。基于4T1细胞模型的体内外研究表明,诊疗试剂38相比对照组(PBS或游离Dox)展现出更强的细胞毒性与更低的副作用,凸显了其在癌症治疗中的潜力。
硝基还原酶响应治疗诊断探针
硝基还原酶(NTR),一种富含黄素单核苷酸(FMN)的酶,在多种肿瘤类型中过表达,其表达量与实体瘤内部的缺氧状态紧密相关,后者主要由血管发育不良或供血不足引起,常见于约50%-60%实体瘤的核心区域,尤其是快速增殖导致的血氧供应受限情况。针对缺氧微环境响应型药物递送系统(DDS)的深入探索已在诸多文献中得到体现。下面,我们将概述分子治疗领域内,乏氧敏感DDS的最新研究进展。
在开发乏氧敏感配方时,面临的首要挑战在于肿瘤的血管生成机制,因其能提升肿瘤区域的氧合水平,进而削弱疗法效果。因此,融合血管生成抑制策略与乏氧响应递药体系,成为提升DDS效能的关键路径。
硝基芳烃化合物作为NTR的理想底物,能够经由NTR催化转化为羟胺与胺类衍生物,且在自催化分解过程中促使芳香环电子重排,释放结合的药物分子。基于此,Kim等人设计了诊疗试剂39(图16),该分子通过硝基苯甲醇连接非甾体抗炎药物吲哚美辛与SN-38。此设计不仅利用COX-2过表达实现肿瘤靶向,还通过吲哚美辛抑制血管生成,增强治疗效果。在模拟缺氧(1%O2)条件下,诊疗试剂39对COX-2阳性A549和HeLa细胞展现浓度依赖性毒性,并在不同尺寸的A549多细胞肿瘤球中显示出与游离SN-38相应的强烈荧光信号,表明其能有效穿透组织并激活药物,可能得益于抗血管生成作用。
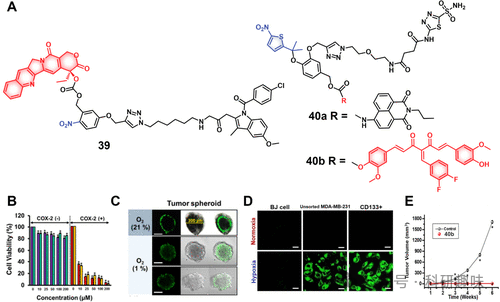 图16.(A)硝基还原酶响应治疗诊断探针的化学结构(39和40)。(B)治疗诊断探针39在不同浓度的COX-2阳性(A549、HeLa)和COX-2阴性(WI-38、BJ)细胞中的细胞活力。(*p<0.05)。(C)用探针39(25μM)处理后,常氧和乏氧条件下HeLa细胞肿瘤球体的荧光图像(第4天)(D)在常氧和乏氧条件(3%氧气)下用40a处理后记录的不同细胞群的荧光图像(比例尺=50μm)。(E)CD+133MDA-MB-231细胞在用DMSO或40b(5.0nM,24小时)处理然后施用于小鼠以测量肿瘤发生后的肿瘤体积与时间图。
图16.(A)硝基还原酶响应治疗诊断探针的化学结构(39和40)。(B)治疗诊断探针39在不同浓度的COX-2阳性(A549、HeLa)和COX-2阴性(WI-38、BJ)细胞中的细胞活力。(*p<0.05)。(C)用探针39(25μM)处理后,常氧和乏氧条件下HeLa细胞肿瘤球体的荧光图像(第4天)(D)在常氧和乏氧条件(3%氧气)下用40a处理后记录的不同细胞群的荧光图像(比例尺=50μm)。(E)CD+133MDA-MB-231细胞在用DMSO或40b(5.0nM,24小时)处理然后施用于小鼠以测量肿瘤发生后的肿瘤体积与时间图。
同一团队进一步研发了针对癌症干细胞(CSCs)的诊疗剂对40a和40b(图16),采用二甲基硝基噻吩替换对硝基苄基作为乏氧响应触发元件,以期凭借其更优的还原电势促进药物释放。治疗部分采用具有抗癌潜能的3,4-二氟亚苄基姜黄素,并借助碳酸酐酶IX(CAIX)抑制剂乙酰唑胺实现对CSCs的选择性靶向。成像方面,则采用萘胺基荧光团。尽管40a和40b在药物释放单元上有所差异,但保留了相似的靶向和乏氧响应机制,故推测两者在细胞摄取和激活特性上具有相似性。通过CD+133MDA-MB-231乳腺癌细胞实验,确认了两者的乏氧敏感激活性能及抗肿瘤活性。特别是,诊疗试剂40b在异种移植小鼠模型中,通过尾静脉注射方式,显著抑制了CD+133MDA-MB-231肿瘤的生长,对比未经治疗的对照组,展现了其治疗潜力。
酯酶响应治疗诊断探针
羧酸酯酶(CES)家族作为一类普遍存在于生物体内的水解酶,专注于催化酯键、酰胺键以及氨基甲酸酯键的水解过程,构成了生物体抵御外来有害物质入侵的重要防线。其中,CES1和CES2这两种同工酶尤为引人注目,受到了科研领域的广泛研究。研究结果表明,与正常组织相比,CES2在病理组织中的表达水平显著升高,特别是在肿瘤组织中尤为明显。
在癌细胞中,过度表达的CES被认为能够显著增强癌细胞的侵袭力、迁移能力、生存能力以及促进肿瘤增殖。例如,在恶性结直肠癌的案例中,CES的活性水平相较于正常组织有显著提升,其中男性患者的CES活性约为正常水平的2-4倍(0.45±0.25U/L对比0.17±0.09U/L),女性患者也呈现出类似的增长趋势(0.45±0.35U/L对比0.12±0.07U/L)。
基于这些发现,科研人员已经开发出了多种针对CES的响应型诊疗试剂,以实现对肿瘤组织的特异性成像和治疗效果的强化。例如,Kunimoto及其团队设计的诊疗试剂41,便是一种通过δ-内酯结构与SN-38经由氨基甲酸酯桥接形成的CES响应性化合物(如图17所示)。这种设计使得在CES的作用下,能够特异性地释放SN-38,从而实现对肿瘤细胞的精准打击。
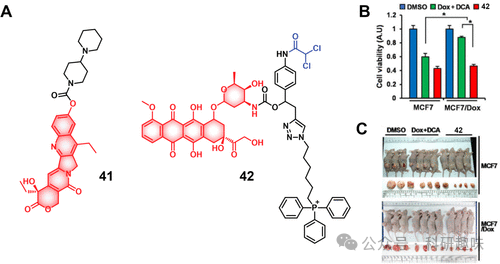 图17.(A)酯酶响应性治疗诊断探针41和42的化学结构。(B)用DMSO、Dox+DCA(1:1)处理后,Dox敏感MCF7和Dox抗性MCF7/Dox细胞的细胞活力或治疗诊断探针42。(C)分别用DMSO对照、Dox+DCA(1:1)或治疗诊断探针42治疗后MCF7和MCF/Dox异种移植肿瘤模型的代表性图像。
图17.(A)酯酶响应性治疗诊断探针41和42的化学结构。(B)用DMSO、Dox+DCA(1:1)处理后,Dox敏感MCF7和Dox抗性MCF7/Dox细胞的细胞活力或治疗诊断探针42。(C)分别用DMSO对照、Dox+DCA(1:1)或治疗诊断探针42治疗后MCF7和MCF/Dox异种移植肿瘤模型的代表性图像。
在临床应用中,化疗面临的主要挑战之一是多药耐药性(MDR)的形成,这种耐药性使肿瘤细胞能在多种化疗药物的作用下存活。据统计,近九成的癌症死亡案例与MDR有密切关系。MDR的形成机制相当复杂,涵盖了药物摄取减少、药物外排泵活性的增强、DNA损伤修复效率的提升、异生物质代谢的加速以及遗传变异(如基因扩增、突变及表观遗传变化)等多种因素。
为了克服这一难题,Kim等人开发了一种新型的诊疗试剂42(如图17所示)。该试剂巧妙地结合了二氯乙酸(DCA)、通过酰胺键连接的阿霉素(Dox)以及疏水性三苯基膦(TPP)线粒体定位基团。其设计原理在于通过恢复癌细胞中异常的糖酵解途径至正常的线粒体氧化磷酸化过程,进而增强癌细胞对化疗药物的敏感性。
DCA作为丙酮酸脱氢酶激酶(PDK)的有效抑制剂,在诊疗试剂42中起到了关键作用。它能够促使癌细胞的糖代谢由糖酵解转向氧化磷酸化,这不仅有助于减少乳酸的积累、降低ATP水平,还能帮助恢复线粒体功能。此外,由于42分子在设计时即考虑到了线粒体定位,因此能够有效规避ATP驱动的ABC转运蛋白介导的初期药物外排,确保药物在细胞内的持久作用。
随着时间的推移,Dox在诊疗试剂42的引导下逐渐转移至细胞核,发挥其抗癌效应。细胞实验显示,诊疗试剂42在A549和HepG2癌细胞中的吸收效果优于正常细胞(如NHDF、IMR90),并且在治疗效果上显著优于单独使用Dox或DCA与Dox的联合使用。
在进一步的研究中,无论是在MCF敏感株、MCF/Dox耐药株还是相应的异种移植小鼠模型中,诊疗试剂42均展现出了显著的治疗活性。这一发现充分证明了其在克服耐药性方面的巨大潜力,为化疗提供了新的治疗策略和方向。
蛋白酶响应治疗诊断探针
蛋白酶,作为催化肽键断裂的核心酶类,在蛋白质降解、消化吸收以及细胞信号转导等生物学过程中占据关键地位。其活性的异常变化与多种病理状态如炎症、肿瘤、心血管疾病以及神经退行性疾病密切相关。鉴于蛋白酶的重要性,近年来科研人员对蛋白酶抑制剂及蛋白酶响应型诊断与治疗探针的研发投入了大量精力。
其中,Caspase家族,特别是caspase-3,作为参与炎症反应、肿瘤发展及细胞程序性死亡调控的关键蛋白酶,备受关注。caspase-3作为一种半胱氨酸-天冬氨酸特异性蛋白酶,可由细胞内在途径或外源性治疗手段(如放化疗)激活,进而引发细胞凋亡。
Byun等人设计了一种创新的Caspase-3响应性抗癌药物43(如图18所示)。这种药物巧妙地将癌症靶向配体Arg-Gly-Asp三肽、Caspase-3识别序列DEVD四肽、酯类敏感连接方式以及化疗药物Dox融为一体。Dox不仅能直接激活caspase-3,而且在caspase-3激活后,通过酯键的水解释放自身,形成正反馈循环,从而显著增强治疗效果。
实验结果显示,该诊疗试剂在整合素阳性的U-87MG胶质瘤细胞中展现出了极高的细胞摄入与毒性,这主要归因于整合素介导的内吞作用。然而,在整合素阴性的HT-29细胞中,其毒性则相对温和。这一自增强凋亡策略在细胞水平上使Caspase-3活性提升了约154倍,并在U-87MG肿瘤小鼠模型中显著抑制了肿瘤的生长,展现了其巨大的应用潜力。
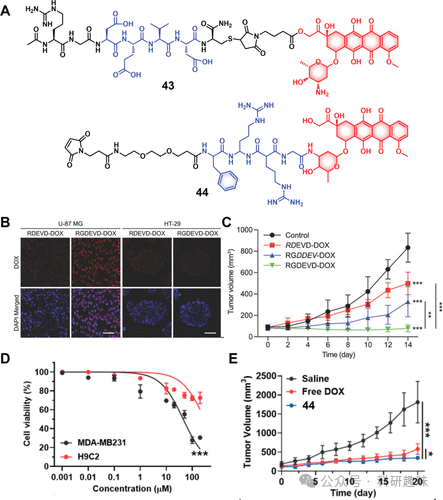
图18.(A)治疗诊断探针43和44的化学结构。(B)用RDEVD-DOX(对照)和RGDEVD-DOX治疗时记录的U-87MG和HT-29癌细胞的共焦图像。红色归因于阿霉素荧光,而蓝色表示细胞核(比例尺=50μm)。(C)分别用盐水、RDEVD-DOX、RGDDEV-DOX或43(RGDEVD-DOX)治疗后U-87MG异种移植小鼠的肿瘤抑制(n=6)(±SD*p<0.05,p<0.01,*p<0.001(与对照相比)。(D)治疗诊断探针44在MDA-MB-231和H9C2细胞中的细胞活力 (E)用盐水、游离Dox或治疗诊断探针44治疗后MDA-MB-231荷瘤小鼠的肿瘤抑制(p<0.05,*p<0.001)。 凝血酶,作为溶酶体蛋白酶家族的重要成员,在蛋白质代谢、免疫调节及细胞生命活动的调控中扮演着关键角色。其表达异常与多种疾病,特别是癌症的不良进展密切相关。特别是Cathepsin B,它在促进细胞外基质降解、加速肿瘤血管生成、侵袭和转移方面发挥着核心作用。Kim等人研发的诊疗试剂44(如图18所示),通过巧妙利用Cathepsin B响应肽序列FRRG,将Dox通过马来酰亚胺分子与白蛋白偶联,实现了药物的稳定化(半衰期显著延长至3.1小时)。在肿瘤微环境中,Cathepsin B的激活能够释放Dox,展现出对MDA-MB231乳腺癌细胞的高选择性毒性。
此外,Tian等人开发的诊疗试剂45(如图19所示)采用了叶酸靶向策略,通过Cathepsin B介导的SN-38递送系统,专一性地针对叶酸受体阳性的癌细胞(如SK-Hep-1、HeLa和Siha细胞)。在细胞内,该系统能够激活并释放活性药物,同时增强细胞核内荧光信号,显著增强了对这些癌细胞的杀伤效果(IC50约2-3μM)。而对正常细胞(如16-HBE)和FR阴性的A549肺癌细胞的毒性有限(IC50=20μM),表明了45在减少SN-38非特异性毒副作用方面的潜力。
组蛋白去乙酰化酶(HDACs)作为核心的表观遗传调节因子,对调控细胞内蛋白质活性与基因表达至关重要。研究指出,HDAC活性的异常增高与多种恶性肿瘤的发生发展,如神经母细胞瘤、胃癌、卵巢癌、结直肠癌及多发性骨髓瘤等存在显著相关性,并常预示患者预后较差。因此,构建针对HDAC的响应性药物递送系统成为了研究热点。Kim等人设计的诊疗试剂46结合了乙酰赖氨酸、Dox和吲哚美辛,旨在靶向COX-2阳性的肿瘤细胞。实验证实,该诊疗试剂在经历HDAC和CTSL的双重酶切后,能有效释放Dox并伴随荧光增强。细胞实验进一步揭示,不同细胞系对46的响应与各自的HDAC、CTSL活性及COX-2表达水平紧密相关,其中HeLa和HepG2细胞展现出最强的治疗效果,并在相应的肿瘤异种移植模型中呈现良好的肿瘤靶向性。
另一方面,Zheng等人提出了一套基于喜树碱(CPT)的HDAC响应型诊疗试剂库47a-c,通过采用多种肽序列如CRGDK、CREKA和TAT,实现对肿瘤的多级靶向策略。这些肽序列分别靶向αvβ3整合素、肿瘤血管和细胞核。实验数据显示,这些诊疗试剂在与HDAC1孵育时,药物释放效率高且快速(如47a在1小时内释放44.1%的CPT),且在无酶条件下保持高度稳定性,体现了高度的酶选择性释放机制。在细胞毒性评估中,所有诊疗试剂在表达HDAC1的癌细胞系中均展示出显著毒性,其中47b在4T1和MDA-MB-231细胞系中表现尤为突出。动物实验进一步证实,这些诊疗试剂能够有效抑制乳腺肿瘤生长,特别是双重组合策略在原位及转移性乳腺癌模型中展现了卓越的抗肿瘤活性。
豆豆蛋白酶(Legumain),作为一种溶酶体/血管天冬酰胺内肽酶,最初在植物中发现,而在哺乳动物体内则主要参与溶酶体功能。它在多种实体瘤中过度表达并与肿瘤进展密切相关,尤其是在前列腺癌、乳腺癌等多种癌症中观察到其活性增强。针对豆豆蛋白酶的活性,Riu等人设计了诊疗试剂48,通过特定的三肽序列Ala-Ala-Asn与Dox偶联,该序列能够被豆豆蛋白酶特异性识别和切割。研究发现,在模拟肿瘤酸性环境(pH 5.5和6.5)下,诊疗试剂48能够高效释放Dox,而中性环境下则几乎无药物释放。共聚焦显微镜分析证实了48通过内吞途径进入癌细胞,实现了增强的细胞毒性和减小的副作用,并通过“旁观者效应”对肿瘤及其周围微环境产生广泛影响。
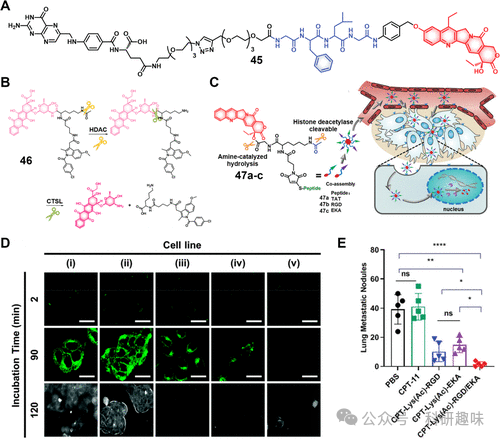
图19.(A)治疗诊断探针45-47的化学结构。(B)示意图显示治疗诊断探针46的激活和药物释放。(C)治疗诊断探针47a-c的化学结构和激活模式,以及它们通过主动和被动肿瘤靶向和细胞核积聚以释放特定药物有效负载而在肿瘤组织中优先积聚。(D)治疗诊断探针46在(i)HeLa、(ii)HepG2、(iii)HCT116、(iv)MIAPaCa-2和(v)Caco-2细胞中的时间依赖性荧光图像。(E)47和对照的抗转移活性显示在实验终点有许多肺转移结节(n=5)。
半乳糖苷酶,作为酶学领域的重要成员,在分解代谢过程中扮演着不可或缺的角色,对生物体的正常运作至关重要。其中,β-半乳糖苷酶(β-gal)作为一种关键的溶酶体水解酶,其主要职责在于从糖共轭物中精准裂解末端半乳糖苷残基,这一独特功能在生物体的代谢调控网络中占据核心地位。
特别值得注意的是,β-gal在多种癌症中呈现异常高表达的现象,这一发现为癌症的靶向成像和药物研发提供了新的策略。近年来,β-gal在癌症诊断与治疗领域的应用研究取得了显著进展。
在诊疗学领域,Kim等人成功研制了一种新型β-gal响应性药物49。该药物通过自巯基连接技术将抗癌药物与半乳糖苷酶分子共价结合,实现了药物的高度稳定性和靶向性。在制备过程中,49在PBS中保持稳定,且不与非特异性受体结合;然而,一旦与β-gal相遇,半乳糖苷酶分子会迅速水解,从而释放出活性药物,并伴随荧光信号的显著增强。
细胞实验结果显示,49能够通过过表达的Asialoglycoprotein(ASGP)受体被高效吸收进入HT-29和HepG2细胞,而在ASGP表达水平较低的HeLa细胞中,其吸收率显著降低。此外,在HT-29和HepG2细胞系中,49展现了浓度依赖性的抗癌效果,显示了其优异的抗肿瘤潜力。
在动物模型中,使用HT-29癌症异种移植小鼠进行的肿瘤抑制研究证实,与单独使用Dox相比,49治疗后肿瘤生长受到显著抑制,这一结果为49作为潜在抗癌药物的开发提供了强有力的实验支持。
此外,Buniya等人也研发了一种新型诊疗试剂50。该试剂将β-gal响应的半乳糖苷酶分子与香豆素荧光团相连,并进一步与吉西他滨耦合。在β-gal的作用下,半乳糖苷酶分子发生水解反应,触发香豆素支架的分子内电子重排,从而释放出吉西他滨,并在短时间内产生强烈的荧光信号。由于细胞内酶对吉西他滨的快速代谢,其半衰期较短;因此,50的策略有望通过减少吉西他滨的不良代谢来提高其治疗效果。
细胞毒性实验表明,与游离吉西他滨相比,50在HepG2细胞中的毒性更高,而在正常HFF-1细胞中的毒性显著降低。这一结果进一步验证了50作为一种新型抗癌药物的潜力和优势。
综上所述,科研人员通过巧妙利用半乳糖苷酶在癌症中的高度表达特性,成功开发出具有靶向性和高效性的抗癌药物,为癌症的治疗领域注入了新的活力,并提供了宝贵的思路和方法。
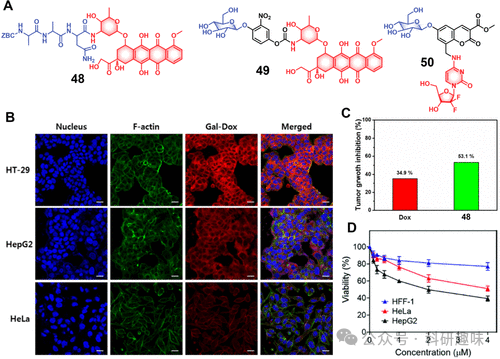
图20.(A)治疗诊断探针48-50的化学结构。(B)用治疗诊断探针49(10μM)处理后的HT-29、HepG2和HeLa细胞的共焦图像。(C)用Dox和探针49处理后HT-29异种移植物(小鼠模型)的肿瘤生长抑制 (D)使用不同浓度的治疗诊断探针50处理后HepG2、HeLa和HFF-1细胞的细胞活力。
【化疗探针】双刺激响应治疗诊断探针
Theranostic Fluorescent Probes
诊疗一体化荧光探针
如前文所述,针对单一刺激(如pH值、谷胱甘肽(GSH)、过氧化氢(H2O2)、酶等)响应性的治疗探针,已成为提升药物效能并降低副作用的可行策略。鉴于肿瘤微环境等复杂病理条件的需求,发展基于双激活机制的治疗探针显得尤为重要,以实现治疗效果与特异性的双重提升。这类探针设计策略既可以涵盖顺序激活机制,以应对不同微环境的梯度变化,也可以实现同步激活,即在同一病灶同时触发多个响应。本章节将概述这两种策略的最新研究进展。
考虑到癌细胞内部的高度异质性,其既可能形成GSH水平升高的还原性微环境,也可能因氧化应激而转变为氧化性状态。这些对立的氧化还原特性不仅在不同肿瘤类型间存在差异,甚至在同一肿瘤的不同部位或单个细胞的不同时间点上交替出现。基于此,Kim及其团队创新性地设计了诊疗试剂51(见图21),旨在通过SN-38的氧化还原敏感传递,实现对这种双面微环境的精准响应。该复合物通过硫醚键将SN-38与COX-2抑制剂吲哚美辛桥接,不仅实现了对癌细胞的定向治疗,还潜在地激发了免疫治疗效应。实验结果显示,在GSH富集的还原环境下,诊疗试剂51经硫醇介导的水解作用释放药物;而在氧化压力下(如H2O2存在时),则形成更稳定的砜/亚砜结构,进而水解脱去活性药物。在体外实验中,51在COX-2阳性的LoVo和SW620细胞系中的摄取和毒性均优于COX-2阴性的细胞(如NHDFs、MCF10A)。动物实验进一步验证,51对结肠癌小鼠模型(SW620)具有显著的肿瘤抑制效果,并伴随着主要炎症标志物(如IL-6、TNF-α、VEGF)的下降,这与诊疗试剂中吲哚美辛的加入密切相关。此外,其他研究亦证实,采用硫醚型氧化还原敏感链接的策略为开发高效治疗药物提供了有力的工具。
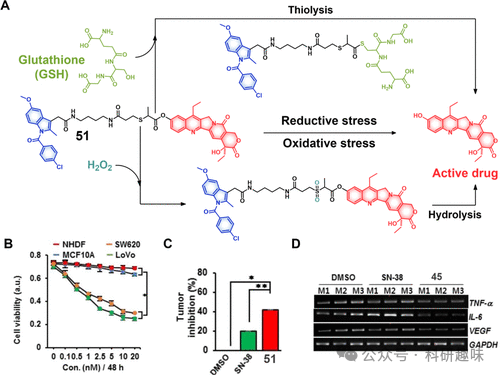
图21.(A)治疗诊断探针51在还原和氧化应激条件下的化学结构和拟议的激活模式。(B)在与不同浓度孵育后观察正常细胞系和癌细胞系中探针51的细胞活力。(C)在SW620异种移植小鼠模型中,DMSO以及等浓度的SN-38和探针51提供的肿瘤抑制作用(n=5,*p<0.05,**p<0.01)。(D)在用DMSO、SN-38和51处理的小鼠组织中观察到的抗炎细胞因子mRNA水平(M1、M2和M3定义了三只小鼠)。 光敏疗法虽以光为媒介调控药物释放与激活提供了契机,但受限于光毒性对周围正常组织的潜在影响,以及日光诱发的非特异性药物泄露问题。为了克服这些限制,Feng及其团队创造性地设计了具有乏氧和光双重响应特性的治疗探针52(图22),通过结合两种激活机制,显著提升了治疗的精准度和安全性。
探针52的设计巧妙运用了4-硝基苄基基团作为其结构的一部分。在常氧条件下(如正常细胞和缺氧的肿瘤微环境),这一基团呈惰性,对光激活无响应。然而,在肿瘤细胞常见的乏氧环境中,4-硝基苄基能够通过硝基还原酶的还原作用转化为中间体52a。此中间态对紫外光高度敏感,经照射后转化为中间体52b,并随后释放活性药物。此过程同时伴随自巯基环化反应,生成发出蓝色荧光的香豆素衍生物。
在MCF-7细胞实验中,当细胞处于模拟的2%氧气缺氧环境中6小时后,经过365nm紫外光照射10分钟,可观察到明显的蓝色荧光发射和增强的细胞毒性。相反,在标准氧浓度(20%氧气)下,未观察到任何荧光信号或细胞毒性反应,这证明了探针52能够精确响应目标癌细胞内的乏氧环境,并实现实时追踪。此外,该设计实现了药物释放的高度空间和时间控制,从而显著提高了治疗的靶向性。
尽管这一设计展现出了巨大的潜力,但将其应用于临床实践仍需进一步的研究和探索。特别是,开发探针52的衍生物,以集成更长波长的光触发机制和红色光谱范围内的荧光报告团,将有助于减少组织穿透障碍并优化生物兼容性。
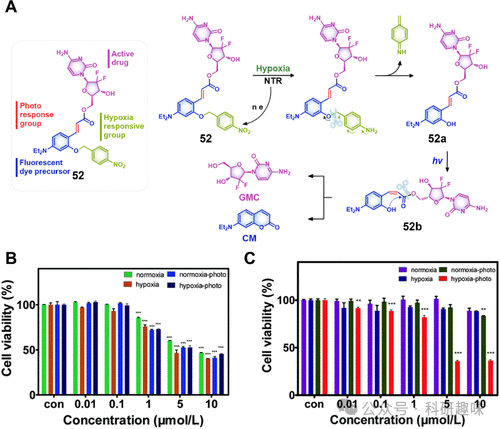
图22。(A)由缺氧和连续光照射控制的治疗诊断探针52的化学结构和激活模式。在存在和不存在光照的常氧和乏氧条件下,用(B)药物本身GEM和(C)不同浓度的治疗诊断探针52处理后,MCF-7细胞的细胞活力。
参考文献
Sharma, A.; Verwilst, P.; Li, M.; Ma, D.; Singh, N.; Yoo, J.; Kim, Y.; Yang, Y.; Zhu, J.-H.; Huang, H.; Hu, X.-L.; He, X.-P.; Zeng, L.; James, T. D.; Peng, X.; Sessler, J. L.; Kim, J. S. Theranostic Fluorescent Probes. Chem. Rev. 2024, 124 (5), 2699–2804. https://doi.org/10.1021/acs.chemrev.3c00778.