【Angew.Chem.】精准调控再进阶!云南大学团队首创“3波长保险锁”:异构体稳定时长突破200小时,100%实现分子开关“按需启动”
✨文章标题:Photoactivatable Hydrazone Photoswitches ✉️作者:Dr. Peng An 等 🔗链接:https://doi.org/10.1002/anie.202521324
[!summary] 总结
研究对象: 开发了一类全新的光活化腙类(Hydrazone)分子开关。
核心机制:
初始状态: 分子以“非活性”的二芳基四唑(diaryl tetrazole)形式存在,就像一把锁死的机关。
第一道光(310 nm): 触发“光激活”。通过氮气释放和分子内亲核加成,将四唑前驱体转化为具备开关功能的腙类分子。
第二、三道光(365 nm / 456 nm): 负责“可逆切换”。一旦分子被激活,这两束光可以控制分子在 E 和 Z 两种异构体之间精准往返,而这两束光绝不会误触未激活的前驱体。
技术亮点:
高度正交: 激活光与切换光互不干扰,实现了真正的时空按需控制。
卓越稳定性: 利用分子的内氢键(N-H...N 和 N-H...O)大幅提升了热稳定性,部分异构体的半衰期长达数月。
应用潜力: 这种“三波长”控制系统在精准医疗、智能材料和催化领域具有极高的应用价值。
在分子机器的世界里,如何实现像精密仪器一样的“即启即停”?2016年诺贝尔化学奖将分子机器带入了公众视野,而分子开关(Molecular Photoswitches) 则是这些微观机器的核心组件。想象一下,如果这些开关在不需要工作的时候能被完全“锁死”,只有在接收到特定的指令信号时才被激活并开始工作,那将为精准医疗和智能材料带来怎样的革命?
近日,云南大学化学科学与技术学院团队在国际化学顶级期刊《德国应用化学》(Angewandte Chemie International Edition)上发表了一项突破性研究。他们设计出一种全新的“光活化腙类分子开关”,通过引入三波长()独立控制系统,实现了分子开关从“静默前驱体”到“活性开关”的按需转换,且异构体的热稳定性提升了数倍,最高半衰期超过200小时。
一、 引言:纳米世界的“影子开关”与它的“死穴”
分子开关是一类能在光刺激下,于两种不同构型(通常是 型和 型)之间往返切换的特殊分子。这种特性让它们在药物递送、生物成像、智能材料及催化等领域大放异彩。
然而,传统的分子开关往往面临一个尴尬的局面:它们始终处于“待命”状态。这意味着,只要光线照射,开关就会切换。但在复杂的生物环境或高精度的工业加工中,我们往往希望分子在到达特定位置或特定时间之前,保持完全的“化学惰性”。
此外,诸如偶氮苯类开关易被还原,或亚胺类开关在酸性条件下易水解等问题,一直是制约其广泛应用的“死穴”。为了解决这些难题,“光活化分子开关(Photoactivatable Photoswitches)”应运而生。这就像给开关加装了一个“保险盖”,只有先揭开盖子,开关才能起作用。
但遗憾的是,在此之前,还没有一种系统能完美实现活化波长与切换波长的完全独立(即正交性)。云南大学团队的这项研究,正是为了打破这一僵局。
二、 核心机制:三道光,锁住无限可能
研究团队巧妙地设计了一种基于二芳基四唑(Diaryl Tetrazole) 的前驱体分子(命名为 O-Tet)。这套系统的运行逻辑如同开启一个三道锁的精密保险柜:
第一道光( nm):开启保险(光活化) 当 310 nm 的紫外光照射前驱体 O-Tet 时,分子内的四唑环会发生迅速的光解,伴随着氮气的释放,产生一种极具活性的中间体——腈基亚胺(NI)。紧接着,分子内预先安置好的“捕获手”——羟甲基(Hydroxymethyl)会迅速出击,发生亲核加成反应,在分子内构建出一个异苯并呋喃腙结构。至此,原本沉默的分子被正式“活化”为具备开关功能的腙类分子(O-Hyz)。
第二、三道光():精准切换(光异构化) 一旦分子被活化,它就变成了真正的“开关”。使用 365 nm 的光()可以将分子推向 型异构体;而使用 456 nm 的蓝光()则能让它快速回到 型构型。
这种设计的精妙之处在于“正交性”: 用于切换开关的 365 nm 或 456 nm 光,能量较低,完全无法撼动前驱体 O-Tet 的四唑环(因为它在 315 nm 以上几乎没有吸收)。这就保证了,只要你不打出第一道 310 nm 的指令,无论你如何变换切换光,分子都稳如泰山,绝不误触。
三、 结果分析:不仅仅是“亮起来”,更要“分得开”
为了验证这一构想,研究团队合成了一系列衍生物(O-Tet-1 到 O-Tet-9),并对其进行了详尽的物理化学测试。
1. 活化过程的高效与纯净
实验结果显示,前驱体 O-Tet-1 在 310 nm 光照下,其在 275 nm 处的特征吸收峰迅速下降,而在 350-400 nm 区域升起了新的峰,这标志着腙类开关的生成。高效液相色谱(HPLC) 分析进一步证实,这种转化是定量且高效的,没有产生复杂的副产物
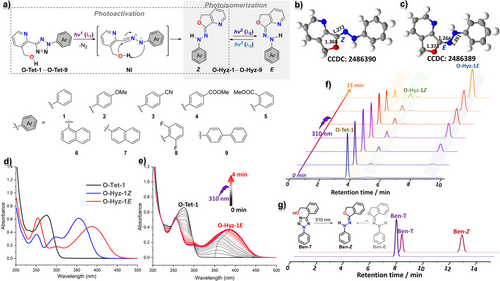
Figure 1 : 展示了光诱导腙类开关生成的全过程,包括 UV-vis 光谱的演变和 HPLC 追踪。可以看到前驱体在 310 nm 下精准转化为活性产物。
2. 令人惊叹的热稳定性:半衰期达 237 小时
分子开关的一个核心指标是稳定性。如果 型还没等光照切换就自动变回去了,那开关就失去了控制意义。 研究发现,通过在分子结构中战略性地引入氮(来自吡啶环) 和氧(来自羟甲基转化后的环),分子内形成了强大的氢键锁。
O-Hyz-1E 的热稳定性极佳,在 25°C 下的半衰期长达 237 小时(约 10 天)。
相比之下,如果去掉分子内的氢键诱导基团(如参考化合物 Ben-T),其生成的异构体会迅速发生热回复,根本无法作为稳定的开关使用。
这一改进意味着,我们可以在更长的时间尺度内,精确控制分子处于哪种状态。
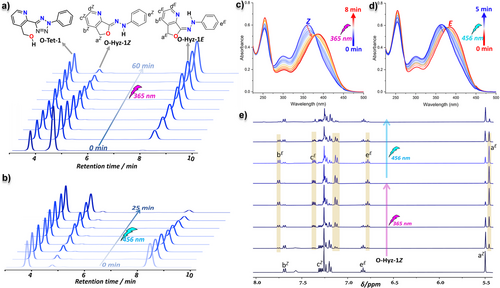
3. 三波长的完美共舞
最令人激动的实验莫过于“三波长门控”演示(Figure 4)。 研究人员选取了性能最优的 O-Tet-5 进行测试:
初始阶段: 10 分钟的 310 nm 光照,将分子 100% 激活为活性腙。
切换阶段: 随后,交替使用 456 nm 和 365 nm 光,分子在 和 态之间反复横跳。
抗疲劳性: 经过 7 个完整循环后,分子的响应信号几乎没有衰减,显示出极佳的耐用性。
更厉害的是“阶梯式活化”实验 27:每次仅给予 30 秒的 310 nm 激活光,分子开关的整体浓度就会像上台阶一样逐步提升,而每一阶内部,分子的异构化切换都保持着高度的独立性和可逆性。
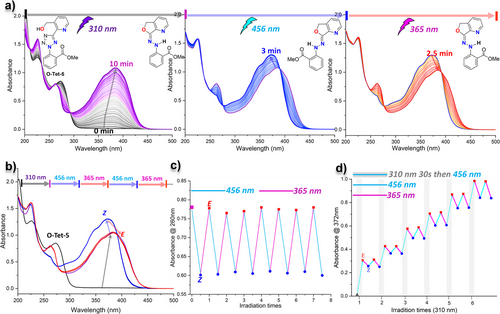
四、 幕后功臣:氢键的“精妙设计”与计算化学的证明
为什么这套系统能表现出如此优异的稳定性?答案藏在分子的微观结构里。
研究团队利用密度泛函理论(DFT) 计算和独立梯度模型(IGMH) 分析,揭示了分子内部的“稳固结构” :
在 异构体中,吡啶环上的氮原子与腙基团的 NH 质子形成了坚固的 N-H...N 氢键。
在 异构体中,新生成的二氢呋喃环上的氧原子则通过 N-H...O 氢键将结构锁死。
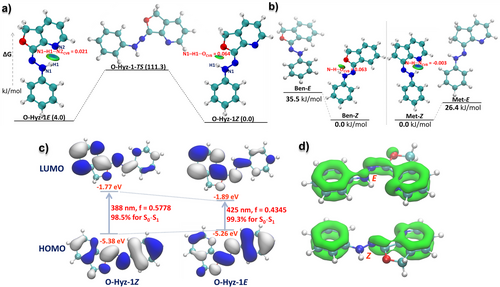
计算数据显示,这种氢键相互作用显著降低了分子的自由能。例如,失去氢键保护的参考结构,其能量比目标分子高出 26.4 至 35.5 kJ/mol。正是这几千焦耳的能量差异,决定了分子开关是在微秒间崩塌,还是在数百小时内屹立不倒。
此外,研究还发现,电子撤回基团(如氰基、酯基) 能显著提升光异构化的效率。在 O-Hyz-5(带酯基)中,光异构化量子产率达到了极高的水平,这为其在低能耗器件中的应用铺平了道路。
五、 总结展望:开启纳米机器的“按需定制”新纪元
云南大学安鹏教授团队的这项工作,成功地将光化学活化与光诱导异构化这两大过程完美耦合,并实现了波长的完全正交控制。
1. 研究局限性与挑战
尽管该系统在实验室环境下表现近乎完美,但要真正走向工业应用,仍有几道坎:
活化波长的红移: 目前的活化波长(310 nm)仍处于紫外区,对生物组织有一定的穿透性限制和潜在伤害。未来能否利用多光子激发或上转换纳米材料将其推向“组织透明窗”的红外区,是值得探索的方向。
水相兼容性: 论文中的测试大多在有机溶剂中完成,而在生理环境(水相、变温、复杂离子)下的表现还需进一步优化。
2. 未来应用场景预测
精准肿瘤治疗(光药理学): 我们可以将前驱体药物注入人体,利用 310 nm 光在肿瘤区域进行局部“解锁”,然后再用可见光精准控制药物活性开关。这样能极大地减少药物对正常组织的毒副作用。
超高密度信息存储: 利用这种三波长控制,可以设计出具有“写入保护”功能的分子存储器。只有经过特定波长“解锁”的存储位才能被修改,极大地增强了数据的安全性。
自修复智能材料: 将这些开关嵌入高分子网络中,可以制造出仅在特定区域发生硬度改变或颜色切换的智能蒙皮。