Visible and near-infrared light-induced photoclick reactions
Abstract
光点击反应结合了光的非侵入性与高时空控制优势以及经典点击化学的特点,在表面功能化、聚合物共轭、光交联、蛋白质标记和生物成像等领域展现出广泛的应用潜力。然而,大多数此类反应依赖于近紫外(UV)和中紫外光来触发,而紫外光可能引发副作用,如细胞凋亡。因此,发展可见光和近红外光驱动的光点击反应系统成为理想的选择。使用更长波长的光不仅可以避免细胞损伤,还能减少光点击试剂及其产物的降解。
为了实现光点击反应向较长波长的转移,研究者们探索了多种策略,包括扩展共轭π系统、三重-三重能量转移、多光子激发、上转换技术、光催化以及设计光电笼等方法。这些方法共同推动了长波长驱动光点击反应的技术进步和未来前景。
引言
化学家们持续探索新的化学反应,旨在简化操作流程、减少副反应、实现精准的化学键形成,并通过直接偶联反应物即时生成大量所需产物。2001年,“点击化学”作为一种革新性的合成方法出现,它极大地促进了这一进程。该方法的核心在于从易于组装的模块化单元出发合成分子。
点击化学反应被定义为具备以下特征:
- 简易操作:反应简单易行,适用于多种底物。
- 高速度与高选择性:反应快速高效,具有优异的选择性。
- 最小或无害副产物:反应显示出极高的原子经济性,产生的副产物极少且无害,并可以通过非色谱法有效去除。
- 环境友好条件:反应宜在室温或温和条件下进行,并使用无害溶剂;若能不使用溶剂则更为理想。
光化学反应因其独特的优势而在满足上述标准方面极具吸引力。光化学与传统点击化学的结合催生了光点击反应,这类反应综合了两者的优点。与传统点击反应中的基态分子不同,光点击反应通过光激活原本惰性的反应物,进而形成新的共价键。这种方法在温和无金属的条件下合成各种有机结构。光子作为非侵入性的反应调控手段,赋予反应无与伦比的空间分辨率和时间控制能力。这些特性使得光点击化学在生物正交转化、表面功能化、聚合物共轭、光交联及生物成像等方面展现出极大的潜力。
Lin等人开创性的工作提出了四唑与烯烃的光诱导环加成反应(NITEC),开启了光点击化学的研究热潮。随后,一系列光点击反应被开发出来,如光引发的硫醇-烯和硫醇-炔反应、叠氮-炔环加成(AAC)、二芳基苯醌-烯光点击反应(photo-DASAC)、氮丙啶连接、9,10-菲醌-富电子烯(PQ-ERA)光环加成反应及酰基硅烷与吲哚的偶联反应等,进一步扩展了该方法的应用范围。
然而,目前多数光点击反应依赖于紫外线(UV)照射,这限制了它们在生物环境中的应用,因为紫外线无法有效穿透生物组织并对细胞有毒。相比之下,可见光(λ = 380-760 nm)和近红外光(NIR,λ = 760-2,500 nm)更适合生物应用和敏感的软材料处理。这些波长较长的光可以有效穿透组织,且散射减少、天然色素(如血红蛋白或黑色素)吸收降低。此外,它们不会引发生物发光,光毒性较低,不会导致生物大分子如DNA、蛋白质、RNA和脂质的有害光化学反应。可见光和近红外光源更加普及,成本更低。因此,迫切需要开发能在生物兼容的可见光和近红外光下激活的光点击元件。
近年来,研究集中于设计可见光和近红外光诱导的光点击反应,包括通过扩展π-共轭系统改变反应物的吸收光谱,设计响应双光子激发的光点击反应物,或者应用上转换纳米粒子在可见光或近红外区吸收并产生用于驱动光加成的紫外光。
接下来,本文将详细概述可见光和近红外光诱导的各种光点击反应及其光激活机制。鉴于硫醇-烯和硫醇-炔光点击反应已有全面综述,本综述将重点介绍其他转化。此外,本文还将讨论针对照射波长进行红移转换的具体策略,并展示由可见光和近红外光驱动的光点击反应的最新进展。最后,本文将探讨该领域面临的挑战、解决方案以及潜在的发展机会。
可见光和近红外光引发的光点击反应的分类
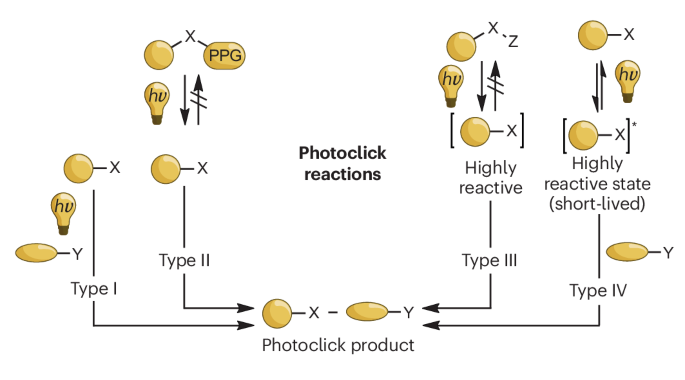
根据光激活的基本机制,本文将可见光和近红外光引发的光点击反应分为四种不同类型():
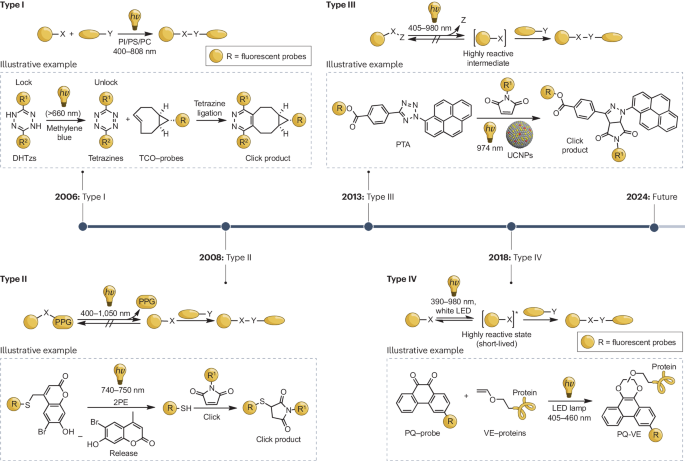
图1.: 可见光和近红外光诱发光点击反应的发展时间表.
点击化学反应根据其对光的不同利用可以分为以下几类:
I 型
这类反应需要光催化剂或光引发剂来激活反应物,进而促进点击反应的发生。示例包括由光触发的叠氮-炔环加成反应(photo-AAC)以及光诱导的四嗪连接(即光激活的反Diels-Alder环加成反应,photo-iEDDA)。
II 型
此类反应为经典点击反应的光激活版本。通过光诱导共价键的断裂,移除一个光可裂解保护基团(PPG),从而形成一个稳定的中间体。该中间体在基态下能与体系中的另一反应物发生点击反应。具体实例有光可释放的硫醇用于硫醇-烯点击化学、光激活的应变促进叠氮-炔环加成(photo-SPAAC)以及光激活的四嗪连接。
III 型
这类反应涉及通过光化学过程不可逆地生成一系列稳定性各异的中间体。在光照条件下,前体分子发生裂解,通常释放出N2或CO2,并产生反应性中间体。这些中间体随后能够被合适的反应伙伴选择性地捕获。典型例子包括光诱导的NITEC、光诱导的邻二甲氧基苯醌的连接、氮丙啶-烯环加成以及光-DASAC。
IV 型
这类反应涉及到短寿命的激发态分子的形成,这些激发态分子可以返回到基态,或者与对应的反应物形成共价加合物。例子包括PQ-ERA以及酰基硅烷与吲哚之间的光点击反应。
I 型光点击反应
在可见光引发的点击化学反应中,当反应物本身不能吸收可见光时,光催化剂或光引发剂的作用就变得尤为重要。在这种情况下,光催化剂或光引发剂有助于生成高活性的瞬态中间体,这些中间体能在较为温和的条件下参与环化反应。
光引发叠氮-炔烃点击反应
铜催化的叠氮-炔环加成反应(CuAAC)是一种重要的点击化学反应,可以高产率和高度区域选择性地生成1,2,3-三唑化合物。这种反应在生物分子偶联、生物标记、药物合成、表面修饰及聚合物合成等领域有着广泛的应用。传统的CuAAC反应通常需要使用抗坏血酸钠等还原剂将Cu(II)还原为Cu(I)。铜和还原剂的存在会立即启动反应,这可能会影响反应的精确控制。然而,借助光化学过程,可以通过激活内源性光活性物质在原位生成Cu(I),实现CuAAC反应的光控催化,并允许对反应进行时空调控。
利用光还原铜络合物的方法主要有两种:直接光还原和间接光还原。在直接光还原反应中,通过特定波长的光照使铜离子还原。在这个过程中,配体吸收光子后,促使分子内的电子从配体的π系统转移到中心金属离子上。这一电子转移将Cu(II)离子转化为Cu(I),同时生成配体自由基。然而,直接光还原通常需要长时间的紫外线照射,这限制了该方法在复杂环境中的应用。相比之下,间接光还原采用一种能够还原Cu(II)离子的光激活剂。在这种情况下,光引发剂在铜络合物吸收光谱范围外吸收光,导致自由基等活性中间体的形成,从而促进Cu(II)还原为Cu(I)。光化学产生的自由基的特性和铜络合物的氧化还原性质对于该过程的成功至关重要。
2006年,Ritter和König首次报道了使用核黄素四乙酸酯和三乙胺(TEA)间接光还原Cu(II)的方法()。他们在实验中将Cu(II)、TEA和核黄素混合后置于含有等摩尔量的炔烃和有机叠氮化合物的溶液中,并暴露于400-500纳米波长的可见光下,结果显示在20分钟内Cu(I)物种催化了CuAAC反应,转化率超过80%。之后,Bowman及其团队开发了一种利用可见光从Cu(II)复合物中原位生成Cu(I)的光化学方法()。他们发现,光引发剂(如Irgacure 819)可以吸收400-500纳米波段的光,从而有效促进Cu(II)向Cu(I)的光还原。通过详细的光谱研究,他们深入了解了这一转化过程和氧化还原特性,并将该系统应用于水凝胶中空间限定的荧光图案的制备。
图 2:光引发的炔吖啶点击反应。
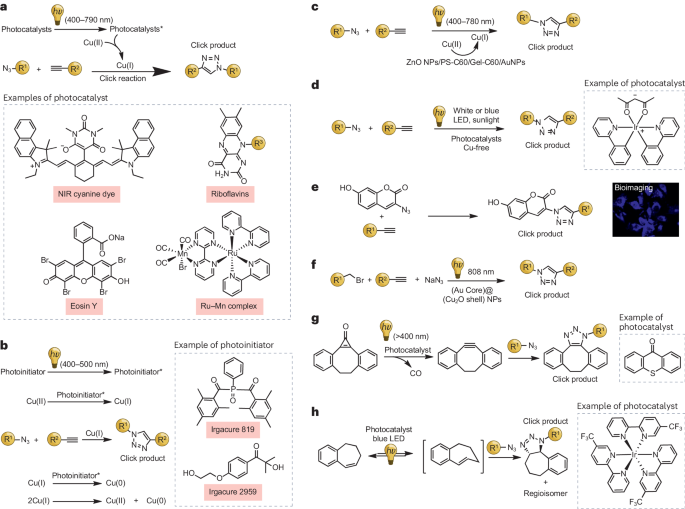
最近,Yagci及其同事展示了富勒烯衍生物(PS-C60和Gel-C60)作为光-CuAAC反应活化剂的有效性,在室温下对400-500纳米波长的可见光表现出高敏感性。这些反应通过光激发和电子传递至Cu(II)进行,操作简便且可直接加入点击成分。此外,该研究组还报道了利用半导体纳米粒子(如氧化锌)作为光催化剂激活CuAAC点击反应的研究,旨在深入理解这类系统的机理,并实现对光引发CuAAC过程的空间和时间控制。
光诱导电子转移(PET)催化剂被用于CuAAC反应中还原铜。相较于传统光引发剂,这些催化剂具有更宽的吸收光谱范围,使得CuAAC反应能够在更长波长的光下进行。2019年,Kutahya等人使用了一种基于近红外花菁染料的PET催化剂,成功实现了使用790纳米波长的光在2小时内催化CuAAC反应。
另一种实现可见光引发CuAAC反应的方法是光氧化催化。2016年,Jain及其同事利用结合了Ru(II)和Mn(I)单元的双金属复合物,提出了一种可见光辅助的光催化CuAAC反应。该复合物展现出显著的催化活性,有效地通过电子传递将Cu(II)还原为Cu(I),进而合成了1,4-二取代的1,2,3-三唑。另外,Wu等人提出了一种高效的区域选择性CuAAC反应方法,仅在白光LED照射下就能达到99%的产率。此方法在空气和湿度下稳定,具有广泛的应用前景,包括在有机合成、药物化学和材料科学领域的应用。
近年来,有机催化剂作为过渡金属替代品在可见光诱导的CuAAC反应中受到关注。2021年,Argüello及其同事成功地将曙红Y或核黄素四乙酸酯作为光催化剂整合入CuAAC反应中。通过稳态光谱和时间分辨光谱研究揭示了反应机制,即三重态的曙红Y可以直接还原Cu(II),而核黄素四乙酸酯则需要光还原。异质纳米铜粒子也成为了有前景的催化剂;例如,Qu及其同事在2020年利用近红外光诱导CuAAC反应与生物相容性纳米催化剂。这种催化系统在活体系统中表现出较高的反应速率和抗肿瘤效果,展示出其在细胞和线虫中的潜在应用价值。
具有局部表面等离子体共振的贵金属纳米粒子能够通过耦合可见光和近红外光子能量来实现光化学反应。Jiang及其同事报告了利用金纳米粒子进行表面等离子体共振增强的CuAAC反应。在空穴清除剂的帮助下,光热加热与热电子诱导的铜(II)还原之间产生协同效应,消除了对抗坏血酸钠还原剂的需求,提高了CuAAC反应的效率。在Au-CuO纳米杂化物的促进下,可见光辅助的光点击反应无需传统的加热或还原剂即可运行。另一种策略是使用等离子金属和半导体组成的异质结构,如Wang及其同事的(Au)@(Cu2O)纳米结构在808纳米波长的光下促进了CuAAC点击反应,产率比热加热条件下高出一倍,实现了1,2,3-三唑的选择性合成。
可见光活化光催化剂也被用于光活化SPAAC反应。Kunishima及其同事介绍了一种使用450-500纳米波长的可见光诱导SPAAC反应的方法。该方法使用光氧化催化剂,使电子从环丙烯酮转移到光催化剂上,生成不稳定的自由基阳离子,从而引发开环并分解为一氧化碳和所需的应变炔。此外,Weaver及其同事开发了一种新策略,利用蓝光激活烷基叠氮化物与苯并环庚烯之间的环加成反应。这一过程涉及由Ir光催化剂介导的苯并环庚烯的光诱导异构化,显示出了高的反应活性和对多种官能团的良好耐受性。尽管这种方法在生物偶联方面非常有效,但存在一些局限性,包括光催化剂在较高浓度时在水中沉淀以及需要较长的照射时间,这可能限制了其在生物系统中的应用。
光催化四嗪-反式-环辛烯点击反应
四嗪限制的烯烃连接,也称为逆电子需求狄尔斯-阿尔德(iEDDA)反应,作为一种生物分子标记技术已广受欢迎。据本文所知,Fox及其团队最早报道了这种反应,其二阶速率常数高达10^6 M^-1 s^-1,是已知最快的生物正交反应之一。然而,这种超快反应速率也带来了局限性,即反应物一旦混合就会立即发生反应,难以实现精确的时空控制。为了解决这个问题,Fox的研究团队开发了一种光化学变体——光诱导四嗪连接(光iEDDA)。
他们采用二氢四嗪(DHTz)作为前体,并使用光敏剂(例如亚甲基蓝),在光照射(λ = 600 nm)下产生单线态氧,后者能够原位氧化DHTz形成四嗪,随后四嗪与反式-环辛烯(TCO)反应形成环状产物。最初的光催化氧化系统在多种应用中均取得了成功,包括激活含有DHTz功能化的蛋白质聚合物纤维用于蛋白质连接。但是,由于亚甲基蓝的光毒性,该系统在活细胞中的应用受到了限制。
为了克服这一限制,Fox及其团队开发了一种基于硅-罗丹明(Si-R)的光催化系统,该系统具有较低的光毒性和良好的水溶性。Janelia Fluor-SiR染料被证实非常高效(通常只需3 mol%的染料),在660 nm波长的光照下,能将二氢四嗪氧化为四嗪,从而促进快速的生物正交共轭反应。使用SiR的红光光催化技术成功实现了透明质酸衍生物在三维前列腺癌细胞培养及小鼠体内的交联,证明了其细胞相容性。
2022年,Fox及其团队进一步拓展了这项技术,展示了SiR光催化系统在亚细胞蛋白质靶标生物正交化学中的应用,实现了明亮荧光团的递送,并且与双光子激发(2PE)结合,即便在内源性蛋白浓度较低的情况下,也能实现实时成像过程中亚厘米级空间分辨率的三维亚细胞结构的模式化。此外,他们还引入了配体导向催化剂和一系列新型光笼,利用适用于体内应用的长波长光来控制药物释放。
除了生物应用外,这种光催化系统还可能用于聚合物材料的功能化。例如,Forsythe及其团队就利用Tz-norbornene iEDDA反应形成了水凝胶。
自从2006年首次报道以来,I型光点击反应已经取得了显著进步。多种光催化剂、光敏剂和纳米粒子(如氧化锌和金)已经被成功用于构建这些系统。I型光点击反应主要包括两种反应类型:光-AAC和光-iEDDA,它们的驱动波长范围从400 nm到近红外光。
尽管近红外光诱导的AAC已经取得成功,但在生物学应用中仍然存在局限性,主要是因为现有系统的水溶性不足和潜在的毒性问题。因此,未来的研究重点应当放在改进这些系统以提高生物兼容性。
相比之下,光-iEDDA系统在红光(高达660 nm)驱动下进行光点击反应,显示出极高的生物兼容性。然而,在未来的发展中还需考虑一些相关因素,例如光点击试剂在生物环境中的稳定性至关重要。尽管TCO是iEDDA反应中的典型亲二烯体,但它在生物条件下的有限稳定性限制了其应用。此外,后续研究工作应着重开发近红外光系统,以进一步提升该系统在体内的性能。预计这一策略性发展将扩大光点击反应在生物医学及相关跨学科领域的影响。
图 3:光活化四嗪_反式_环辛烯点击反应。
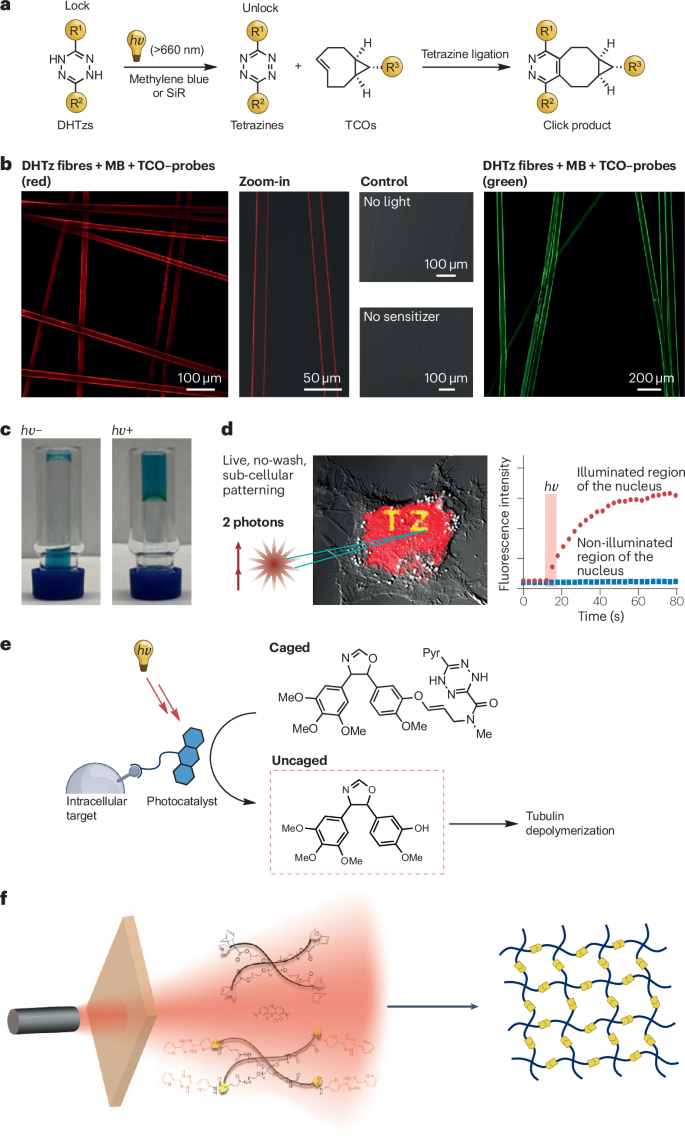
a,光 iEDDA 反应。b,用反式环辛烯(TCO)探针处理活化的 DHTz 纤维 1 分钟并冲洗后的共焦图像。c,红光光催化开启Tz结扎以形成透明质酸水凝胶。在光照射前(hν-)和光照射后(hv+)进行的小瓶倒转测试表明了从液体到水凝胶的转变。观察到的蓝色是由于 SiR 染料造成的。d,左图显示的是活细胞,在双光子激发(880 纳米)下细胞核上的免洗光图案,右图显示的是细胞核被照亮区域(左图中的黄色区域)在 633 纳米波长下的荧光强度。e,与远红光结合的亚细胞局部光催化。f:通过光 iEDDA 反应使聚合物材料功能化。SiR,Si-罗丹明。b、d和e部分经授权改编自参考文献。ACS。第c和f部分经参考文献授权转载。部分c和f分别经 ACS 上的参考文献许可转载。
第二类光点击反应
可光照释放的硫醇-烯光点击反应
光电笼(PPG)可以通过遮蔽硫醇的点击功能来实现对硫醇-迈克尔加成反应的光控。当硫醇被光化学释放后,硫醇-烯点击反应便能正常进行。需要注意的是,裂解掉的PPG会作为副产品保留在反应混合物中。这一点与其它光点击反应不同,后者通常不会释放副产物或者只会释放极少量的副产物(例如四唑-烯反应中的N2、茚酮-烯反应中的CO2或基于环丙烯酮的光-SPAAC反应中的CO)。
尽管如此,这类反应主要用于大分子(如聚合物或生物大分子)和表面修饰,在这些应用中,PPG衍生的副产物相对于点击柄而言体积较小,因此可以容易地去除或被接受。
在这些应用中,最常用的光保护基团是_正_硝基苄基及其衍生物。然而,这些基团的应用存在一些缺点,比如需要高能量的紫外光、产生有毒且强烈吸收光线的副产物以及底物释放效率低(光反应量子产率低(_Φ_P))等问题。相比之下,Furuta等人发现的香豆素光电开关由于能够在可见光触发下高效释放底物,并且能够使用双光子技术而受到青睐。香豆素衍生物经历一个解笼过程,在此过程中,C-X(X = N、O、S)键受到光激发并振动弛豫至S1状态,随后发生异裂。这会产生由甲基香豆素阳离子和阴离子离去基团X-组成的离子对中间体。离子对分离后,通常会被亲核溶剂捕获,形成稳定的产物,并释放出官能团。键的断裂和随后的光释放效率取决于单离子离子对的稳定性,而在香豆素环上引入电子给体基团可以增强这种稳定性。通过加速异质分解可以减少重组的发生,使用碳酸酯连接替换不良的离去基团可以提高释放效率。
图 4:可光释的硫醇-烯光点击反应。
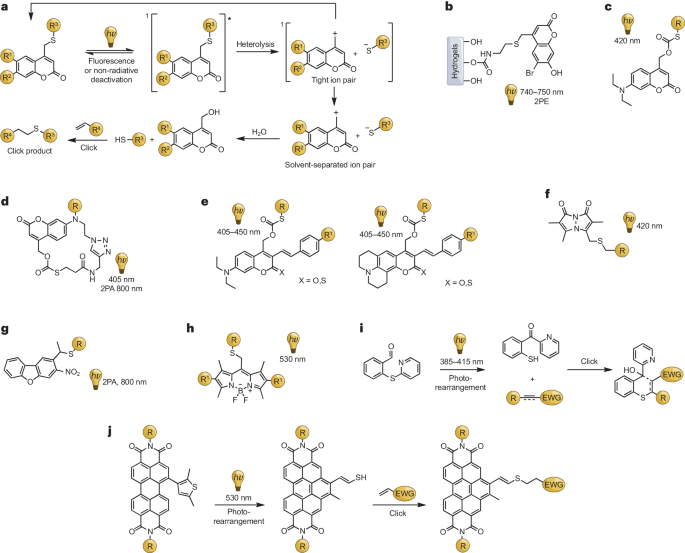
a,可光释的硫醇-烯光点击反应的拟议机理,以香豆素衍生的光点击反应为例。b-h,可见光和近红外光激活光引发剂的化学结构。i,可见光触发硫醇从_正_-硫代吡啶基苯甲醛重排过程中释放出来,用于硫醇-迈克尔反应。j,基于光环化和重排过程的 “免清洗 ”绿光触发的过烯双亚胺骨架硫醇释放。2PA,双光子吸收;2PE,双光子激发。
2008年,Shoichet及其团队展示了香豆素类化合物在双光子激发(2PE)过程中的潜力。他们使用6-溴-7-羟基香豆素衍生物对琼脂糖进行了共价修饰,制备出一种水凝胶,其中的硫醇基团可在紫外线或红光激光激发下被解开(图4b)。这使得生物素等分子得以精确固定,形成化学图案化的水凝胶,可用于调控组织工程中的细胞行为,突显了香豆素化合物在双光子吸收(2PA)研究中的潜力。
2014年,Lin及其团队通过引入7-氨基香豆素分子,推动了单光子激活、可见光触发的光点击反应的发展,使硫醇基团能在420纳米光的照射下得到可控释放。这有助于原位生物共轭反应,构建生物功能材料,如蛋白质-聚合物和量子点-聚合物混合物。他们进一步开发了一种三维细胞培养基质,利用405纳米和800纳米光实现了硫醇-迈克尔加成的时空可控性。该团队还推出了一类新的香豆素光笼,其富含电子的苯乙烯基团具有长波长吸收、大的双光子吸收截面和高的解笼量子产率。这些光笼被用来构建具有独特光漂白特性的光交联水凝胶,克服了内部滤光效应,适用于高浓度或厚样品。此外,他们还合成了一种光致伸缩性聚合物,即使在增加水凝胶厚度的同时也能实现快速凝胶化,并降低了光照射时的强度衰减。
除了使用香豆素类PPG外,还开发了一系列用于硫醇光致化的创新策略,为不同的应用提供了多功能性。溴比曼作为一种可见光响应型光致变色基团被引入,它具有出色的水溶性、生物环境中的稳定性和低细胞毒性。在碱的存在下使用溴代双烷基,可实现高效的硫醇光诱导,并可应用于荧光蛋白标记。这种方法可以在420纳米可见光下促进多臂聚乙二醇(PEG)与多种迈克尔受体的功能化交联聚合。这种技术可以快速释放阴离子硫醇,在30分钟内与生物环境中的迈克尔受体自发反应,无需额外的催化剂。
硝基二苯并呋喃(NDBF)是一种有效的含硫肽保护基团。Distefano及其团队通过紫外光(365纳米)或近红外光(800纳米,2PE)照射实现了高效的硫醇脱保护。Shoichet及其团队利用与基质金属蛋白酶可清除肽交联的NDBF共轭透明质酸水凝胶创建了一个三维乳腺癌侵袭平台,展示了通过双光子辐照固定马来酰亚胺修饰的生物分子。
受硼-二吡咯并二酮(BODIPY)分子衰变为基态离子对的启发,Truong及其合作者探索了在绿光(530纳米)下进行亲核硫醇-炔加成的BODIPY光笼。这种创新方法得益于BODIPY光载体在环境光下的优异稳定性,有望在绿光激活连接之前用于材料储存和加工。
Barner-Kowollik及其团队展示了一种光重排策略,即用可见光(385-415纳米)诱导_ortho_-硫代吡啶基苯甲醛重排,从而产生活性硫醇。这种硫醇与缺电子的炔烃或烯烃发生自催化、无添加的硫醇-迈克尔加成反应。Zhang等人利用过二亚胺(PBI)分子扩展了这一策略,证明了在可见光(530-560纳米)照射下发生的两步光诱导环化和重排反应。从PBI分子中释放出的反应性硫醇可成功实现硫醇-烯连接,为利用与组织相容的光进行深层组织穿透成像的原位生物标记带来了潜力。这些进展有助于扩大硫醇释放控制工具箱的应用范围。
光激活应变促进叠氮-炔环加成反应
尽管铜催化的叠氮-炔环加成(CuAAC)反应是一种广泛应用的技术,但在生物应用中其使用受限于对铜毒性的担忧。为了解决这个问题,Bertozzi 及其同事提出了应变促进的叠氮-炔环加成(SPAAC)的概念。这项创新技术开发出了多种受约束的炔烃,例如氟化环辛炔、二苯并环辛炔以及硫代环炔烃。这些特殊的炔烃能够进行快速且无需铜催化的叠氮-炔环加成反应,为化学转化提供了一种高效且多用途的方法。先前的研究已经表明,环丙烯酮在紫外线照射下会分解生成炔烃,并释放出一氧化碳分子。环丙烯酮的高环应变特征使其能转化为能量更有利的环辛炔应变三键。这种转化允许受约束的环炔在光的诱导下脱保护,并自发参与叠氮-炔环加成反应。
基于早期研究,Popik 及其同事引入了一种光诱导反应,从受光化学保护的环丙烯酮中生成二苯并环辛炔。由此产生的受约束炔烃在无需催化剂的情况下进行叠氮-炔环加成反应,生成三唑。这种方法利用染料功能化的炔烃实现了活体系统中生物分子的可视化,并有望为微阵列制备提供精确的时间和空间控制。然而,所需的紫外线限制了其更广泛的生物应用。
2016年,同一研究小组利用一种新型光化学前体——二苯偶氮(λ = 420 纳米)解决了紫外线限制的问题。尽管存在明显的环应变和反芳香性结构,二苯并二噁英在水溶液中依然表现出了卓越的稳定性,为蛋白质功能化提供了一个多功能平台。可见光激活的 SPAAC 被证实有效,从而扩展了生物正交标记的应用领域。随后,该研究小组进一步推进了光-SPAAC 的应用,展示了环丙烯酮笼型二苯并环辛炔的非共振双光子和三光子激发(分别对应 690 纳米和 1,050 纳米)。这一突破使得近红外光控 SPAAC 成为可能,减少了紫外线的吸收、散射和光毒性,实现了更深的组织穿透。多光子触发提供了亚微米分辨率的精确三维控制,是实现在特定细胞或亚细胞器内进行生物分子衍生化的重要工具。
图 5:光活化菌株促进的叠氮-炔环加成(photo-SPAAC)和四氮连接。
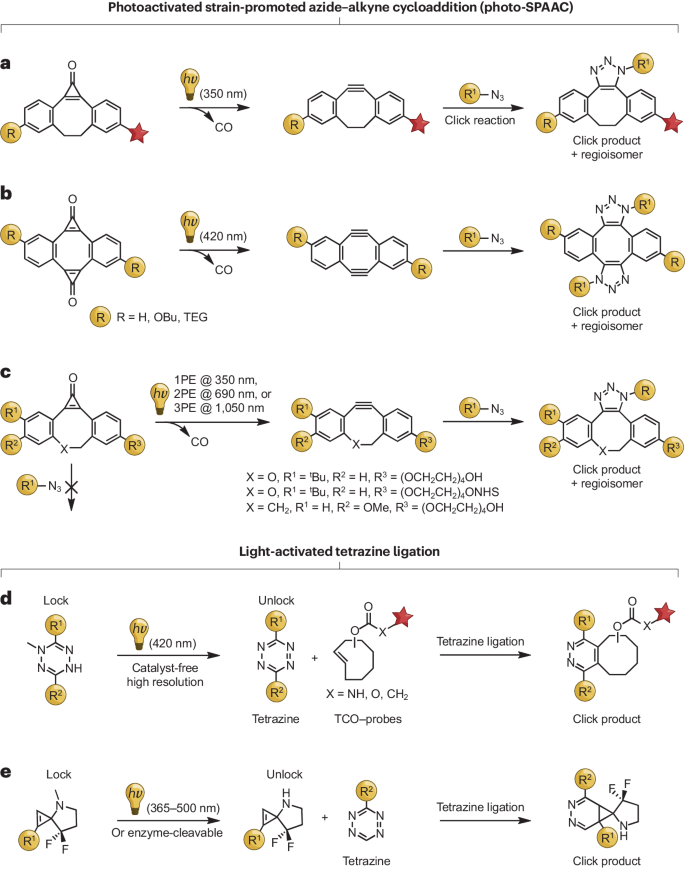
a,无金属叠氮到乙炔环加成反应的光触发。红星代表荧光探针组。b,可见光激活的 SPAAC 双环化反应。c,单光子活化和多光子活化 SPAAC 反应。d*,光激活的光保护 DHTz的解笼导致四嗪的形成,四嗪可与反式_环辛烯(TCO)等二烯烃快速反应。E*,用于控制环丙烯-Tz生物正交连接的多样化和模块化笼式策略。1PE,单光子激发;2PE,双光子激发;3PE,三光子激发。
光激活四嗪连接
四嗪与受约束的二烯烃之间的生物正交环加成反应因其快速反应速率而在标记蛋白质、脂质和糖类方面极具潜力。然而,在哺乳动物活细胞中实现这类反应的精确时空控制一直是一个挑战。Devaraj 及其合作者近期的研究解决了一个关键问题,即如何在活体哺乳动物细胞内控制此类生物正交环加成反应(见图)。他们采用了光敏化的 DHTzs,在可见光(λ = 405 纳米)的照射下,DHTzs 能够转化为活性四嗪,从而与 TCO 等二烯烃进行快速、可控的环加成反应。光诱导条件下 DHTzs 的稳定性使得它们可以预先整合进生物大分子中,用于单细胞修饰和潜在的光诱导治疗。
另外,Laughlin 及其同事开发了一种环丙烯-Tz 连接策略,通过 PPG 屏蔽环丙烯的反应性(见图)。他们设计了一种特殊的笼状螺环丙烯衍生物,其中含有一个大的光保护基团来阻止环化反应的发生。在光照(365-500 纳米)作用下,保护基团被移除,释放出未受阻碍的环丙烯,使之易于与四嗪发生反应。这一策略为实现所需化学修饰提供了一种精确而可控的方法。
光点击反应的第二类,通常被称为光激活点击反应,主要包含三种类型:(1) 光释放硫醇-烯光点击反应;(2) 光-SPAAC;(3) 光激活四嗪连接。这些反应的驱动波长范围从 390 纳米至近红外区域,最远可达 1,050 纳米。值得注意的是,在光-SPAAC 系统中,生成的环辛炔可能会与细胞内的硫醇(如谷胱甘肽或蛋白质中的半胱氨酸残基)快速反应,这限制了其在生物环境中的适用性。此外,光活化过程中释放出的结构化合物,如香豆素和 BODIPY,与传统意义上的点击反应定义有所偏差。这些稳定化合物在体系内的扩散也会影响光点击反应的空间和时间精确度。
第三类光点击反应
光引发的腈亚胺介导的四唑-烯环加成反应
起初,高活性腈亚胺是通过四唑在高温(>150 °C)下的热分解获得的,该过程释放出 N2。同样,这一分解过程也可以通过光照实现,且光照波长需要与四唑上的发色团相匹配。光化学生成的腈亚胺随后与烯烃发生 1,3-二极环加成反应,形成共轭的五元吡唑啉环作为光点击产物。这一过程被称为 NITEC(腈亚胺介导的四唑-烯环加成),生成的吡唑啉通常具有荧光特性,类似于 PQ-ERA 反应,可用于实时监测反应进程。尽管多种类型的烯烃都可参与该反应,但那些带有共轭或电子吸引性取代基的烯烃表现出更强的偶极性。生成腈亚胺的一个优点是唯一的副产品是惰性的氮气。NITEC 反应与多种溶剂(包括水介质)兼容。近年来,NITEC 反应因其良好的动力学特性和生物相容性,在表面功能化、聚合物共轭、蛋白质标记以及细胞成像等领域得到了广泛应用。
2015 年,Barner-Kowollik 及其团队采用相似的策略,在 420 纳米波长下实现了芘四唑的可见光触发 NITEC 反应。这种光点击反应在小分子连接和聚合物改性等方面展现出了出色的反应活性。他们进一步开发了一种对绿光响应的二甲基氨基芘芳基四唑,能够在 515 纳米波长的光下被激活,成为理想的 λ- 正交反应体系。四唑在不同波长光照射下的显著光活化能力促进了 NITEC 与其他光点击反应的整合。近年来,人们对于 λ- 正交光点击表面功能化策略的兴趣逐渐增加。这些策略涉及在存在其他可光活化的官能团的情况下,选择性地激活可光活化的四唑,从而实现对表面改性的精确控制。2018 年,Barner-Kowollik 团队取得了突破性进展,在 NITEC 反应中成功运用了 λ- 正交光活化,通过不同波长的光(紫外光:320 纳米;可见光:410-420 纳米)进行分层图案化处理,实现了复杂的表面修饰。利用飞行时间二次离子质谱 (ToF-SIMS) 对该无掩模光刻技术在硅晶片上的应用进行了可视化展示,显示了其巨大的潜力。
2013 年,An 等人采用 π-系统共轭延伸策略设计了一种四唑,该四唑可通过 405 纳米激光激活用于 NITEC 反应体系。这些四唑展示了非常高的光点击反应活性(k2 高达 1,299 ± 118 M⁻¹ s⁻¹)和更高的光产率(ΦP = 0.16),这两项指标均超过了先前的体系(ΦP = 0.006-0.04)。使用水溶性的四氮唑-烯系统能够选择性地标记并成像 CHO 细胞中的微管,并具备空间控制能力。通过在 C5 位引入扩展的 π-共轭,可以提供红色荧光读数,这对于研究生物系统内部极性变化具有重要价值。
Lin 及其团队利用双光子激发 (2PE) 的优势,在 NITEC 光点击反应中实现了出色的空间和时间控制。他们使用波长为 700 纳米的飞秒脉冲激光触发反应,展现出 0.067 ± 0.001 μM min⁻¹ 的零级动力学 (_k_0)。四唑在萘的 β 位上含有助色和聚乙二醇基团。这种由 2PE 促进的 NITEC 反应成功应用于活体哺乳动物细胞内微管的空间控制显微成像。此外,还使用了上转换纳米粒子 (UCNPs),使 NITEC 反应能够使用红移光(974 纳米近红外光)。不同于 2PE,UCNP-NITEC 系统通过发射的紫外光子促进反应。Zhang 及其团队通过将 UCNPs 与四唑共价连接,开发了一种近红外激活的上转换纳米探针(四唑-UCNP),进一步提升了效率。该系统有助于在小鼠特定肿瘤组织中实现硅酸的空间选择性可视化,为生物医学研究提供了一种可控的标记策略。
图 6:光引发的腈亚胺介导的四唑-烯环加成反应(NITEC)。
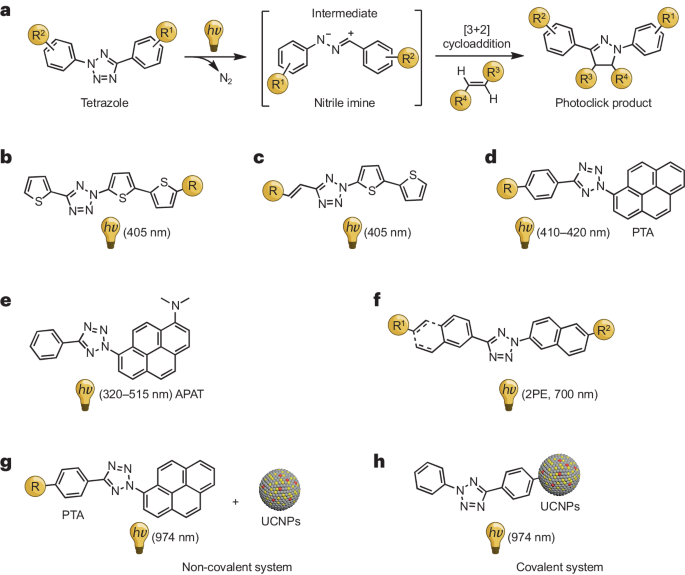
a,NITEC 光点击反应的拟议机理。b-h,可见光和近红外光活化四唑的化学结构。2PE,双光子激发;APAT,二甲基氨基芘芳基四唑;PTA,芘芳基四唑;UCNPs,上转换纳米粒子。
光诱导 ___-奎宁二甲烷的连接
-奎宁二甲烷(-QDMs)在促进 Diels-Alder 加合物形成方面展现了优异的能力(图)。这些系统的应用广泛,涵盖了表面改性、生产序列定义的大分子、聚合物-聚合物共轭以及三维激光写入等领域。
图 7:光诱导的___-二甲基喹啉(___-QDMs)连接和氮丙啶-烯环加成。
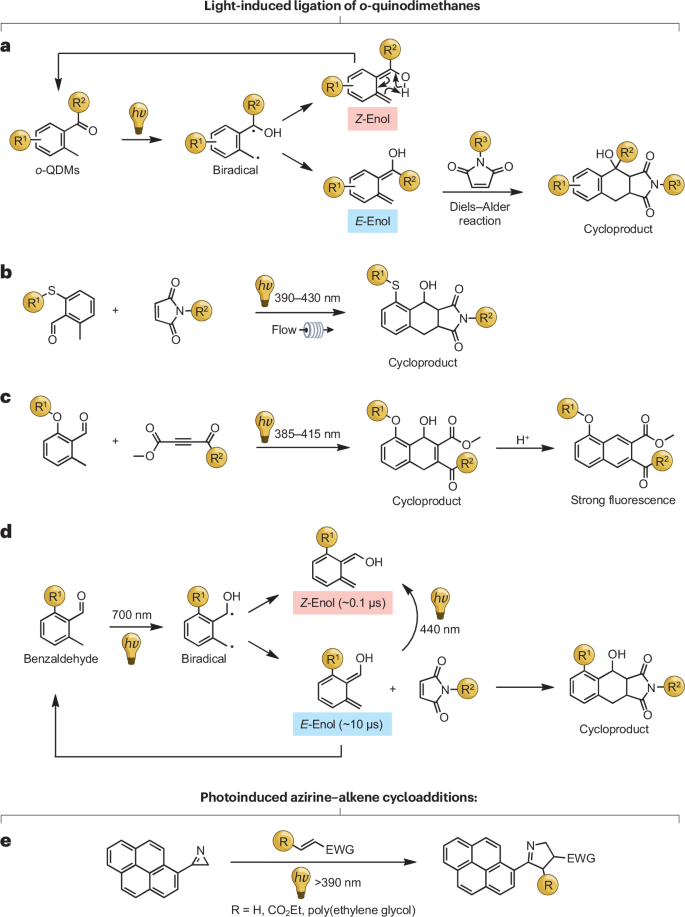
a,光诱导 o-QDM- 烯反应的拟议反应机理(以马来酰亚胺为例)。b,可见光诱导的 ___-QDM-硫醚反应。c*,可见光诱导的 o-QDM 与门控荧光自报告的连接。d,近红外光诱导 o-QDMs 连接的拟议反应机制。e*,可见光诱导的氮丙啶-烯环加成反应。
Barner-Kowollik团队的研究:___-QDM与二烯烃间的光诱导点击反应
Barner-Kowollik及其同事研究了___-QDM与二烯烃之间的光诱导点击反应,重点关注表面官能化和聚合物-聚合物共轭的波长依赖性。他们报道了一种使用活性___-QDM硫醚和缺电子烯烃,在可见光(390-430纳米)下诱导的Diels-Alder转化(图)。通过在___甲基苯甲醛中进行氧硫置换来生成反应性的二烯烃。荧光产物经过受控的E1消除后迅速转化为荧光萘,这使得能够进行原位荧光监测。2017年,Wegner等人展示了一种协调的光控制策略,在700纳米光照射下,_甲基苯甲醛通过双光子吸收产生_E-和_Z-烯醇。其中,寿命较长的_E_形式参与了Diels-Alder反应,而寿命较短的_Z_形式则效率较低。同时,440纳米波长的光照可以使_E_异构体转变为_Z_异构体,利用受激发射耗尽实现了空间限制,并在激光写入图案中达到了亚衍射极限分辨率。
光诱导氮丙啶-烯环加成反应
光解2_H_-氮丙啶会产生高度活性的腈酰亚胺中间体,这些中间体能够与多种二烯烃进行环加成反应,形成环加成产物。2010年,Lim和Lin首次采用氮丙啶连接技术,在水溶液环境中通过氮丙啶-烯环加成反应高效修饰了蛋白质。原位生成的腈酰亚胺表现出极高的反应性,因此这种方法也被应用于缺电子烯烃。在此基础上,Barner-Kowollik及其同事通过引入芘分子进一步扩大了氮丙啶连接技术的应用范围,实现了在可见光(>390纳米)下与多种缺电子烯烃的反应。这一系统在有机合成和聚合物连接领域展现出了高效性,并且能够在短时间内完成选择性、清洁及完全端基的修饰。
Diarylsydnone-alkene 光点击反应
茜酮因其独特的双极性分子结构——同时带有正电荷和负电荷并在环上分布——而展现出特殊的物理和化学性质,这使得它们成为合成各种杂环体系的有前途的前体。2018年,Yu及其同事开发了一种创新的可见光诱导二芳基茜酮(DASyd)-烯环加成法(图)。这种方法被称为光-DASAC,它涉及合成一系列DASyds,并在光诱导的1,3-二极环加成反应中与各种烯烃表现出优秀的反应活性。他们还展示了一种用于TCO蛋白质的光诱导荧光标记技术,这表明了该方法的多功能性和广泛的应用潜力。
**图 8:Diarylsydnone-alkene 光点击反应(photo-DASAC)。
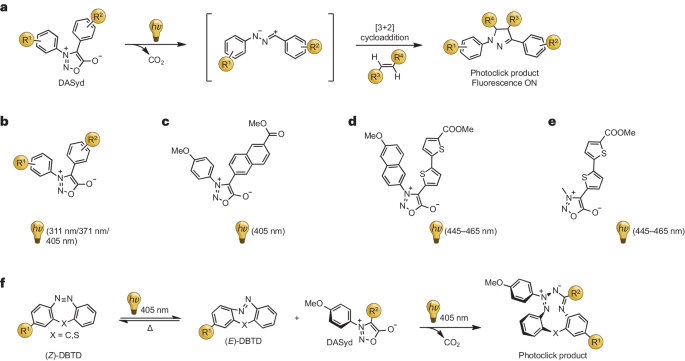
a,二芳基萘-烯光点击反应的拟议机理。b-e,可见光激活的二芳基萘酮(DASyds)的化学结构。f,DASyd-二苯并硫氮杂卓(DBTD)光点击反应的反应方案。
随后,同一研究团队在405 nm的光照条件下,展示了DASyd与应变性炔烃(如双环炔基)之间的快速光点击反应,并通过选择性标记细胞表面蛋白证明了其生物相容性(图)。进一步的研究中,他们开发了一种新型的两亲性极性化合物——二苯并噻二唑(DBTD)。可见光触发了DBTD的异构化过程,从而增强了其在环化反应中的活性。E-DBTD异构体的反应速率是_Z_-DBTD异构体的6.6倍,这使得蛋白质和细胞的快速荧光标记成为可能(图)。
为了解决DASyds仅能在405纳米范围内被激活的问题,Yu及其同事通过在特定位置引入环烷或低聚噻吩来扩展发色团的吸收光谱,产生红移效应(图)。这一策略使得化合物能够在更长的波长(445-465纳米)下被激活,潜在的应用包括在单细胞水平上实现药物的光释放和追踪。
第三类光点击反应是最受欢迎的类型之一,已经历了广泛的开发和应用。这类反应系统包括四个关键反应:(1) NITEC;(2) ___-QDM的光诱导偶联;(3) 光诱导氮丙啶-烯环加成;(4) 光-DASAC,它们的激活波长范围从390纳米延伸至近红外区域。
NITEC反应和光-DASAC反应因其出色的反应活性和良好的生物相容性而受到广泛关注,并已被应用于生物成像和生物大分子的修饰。尽管取得了这些进展,但在体内应用这些光化学转化仍较为有限,需要进一步改进。
相比之下,其他两种反应的应用主要集中在聚合物合成、表面改性和光图案化方面。为了拓宽这些反应的应用范围,还需要在其他领域进行更多的探索和开发。
第四类光点击反应
PQ-ERA 光环加成反应
PQ-ERA 反应的机理是利用紫外线至蓝光范围内的光来激发 PQ(图)。激发后,基态 PQ 进入 S2(1_ππ_)态,随后经历内部转化形成最低激发态 S1(1_nπ_)单态。接下来,发生高效的系统间交叉过程,形成两个最低三重态:T1(3_ππ_)和 T2(3_nπ_)。值得注意的是,T1 是 PQ-ERA 反应的反应态(图),而 T2 也占有一定比例。这些三重态的能量非常接近。然而,由于需要强汞灯作为光源,且反应时间长(需数天),这在过去的 70 多年间一直阻碍着 PQ-ERA 在合成中的进一步应用。
2018 年,Zhang 及其同事利用高功率 LED 光源解决了 PQ-ERA 光点击反应中的某些难题,展示了该反应在 5 分钟内即可完成,并且即使在水环境中也能获得高达 93% 的光加成物(图)。他们通过在活细胞中标记细胞膜,展示了 PQ-ERA 光点击反应的生物相容性,显示了其在生物应用方面的潜力。Feringa 及其同事展示了 PQ 单元与生物大分子(如万古霉素和前列腺特异性膜抗原抑制剂)的结合,扩大了其在各种生物环境中的应用,尤其是用于正电子发射断层扫描成像的放射性标记(图)。
2022 年,Zheng 等人引入了 PQ-ERA 生物正交环化技术,促进了荧光探针的构建(参考文献)(图)。这种方法可以利用可见光(450 纳米)高效标记和可视化寨卡病毒,为理解病毒的致病性提供了有价值的工具。Xie 及其同事提出了顺序光照射-正交报告标记法,这是一种通过引入代谢前体引发的生物正交反应,能够以空间和时间分辨率对活细胞中的细胞表面生物分子进行可控标记(图)。
PQ-ERA 光环加成法的高选择性可与四氮唑-烯光解法并行应用,从而在水凝胶基质中实现生物活性分子的 λ 正交连接。Wu 等人利用这种方法制造了双层网络水凝胶,可以进行空间分辨的生长因子修饰(图)。2020 年,Adronov 及其合作者利用可见光对共轭聚合物骨架进行功能化处理,在材料科学领域取得了突破性进展,创造了首个可光照图案化的共轭聚合物骨架,为开发包括光响应电子器件和传感器在内的先进材料提供了可能性(图)。
最近,PQ-ERA 光点击反应不仅被广泛应用于各个领域,而且在提高其可调性方面的研究也引起了广泛关注。Feringa 及其同事探索了 PQ-ERA 光点击反应的精确控制和微调可调性,研究了取代基对反应双方的影响(图)。他们发现,使用烯胺作为 ERA,可以显著加快转化速度,提高光反应量子产率、多色发射输出和加合物的高荧光量子产率。该反应在 390、420 和 445 纳米等多波长光下表现出很高的效率,从而可以在溶液中和纳米粒子表面进行超快化学选择性官能化。
为了提高效率,同一研究小组在 PQ 核心引入了一个噻吩分子,增加了反应性三重态的数量。这种修饰使得 PQ-ERA 反应在可见光照射下具有极高的光反应量子产率(高达 98%)、高二阶速率常数(高达 1,974 M⁻¹ s⁻¹)和显著的耐氧性,从而在环境条件下实现了接近定量和快速选择性的点击反应(图)。
PQ-ERA 光点击反应虽然已成功应用,但由于其依赖于高能紫光或蓝光,可能会产生不必要的反应性,因此带来了挑战。为了克服这一问题,Feringa 及其同事采用了三重三重能量转移 (TTET) 工艺(使用 DiIbodipy 作为光敏剂),将激发波长转移到绿光(530 纳米)或橙光(590 纳米)(图)。与一般 PQ-ERA 系统相比,这种双波长可见光诱导的正交光点击反应系统的波长偏移超过 100 纳米。该反应显示出快速的动力学(_k_2 ≈ 87.9 M⁻¹ s⁻¹)和高化学选择性,即使存在催化量的光敏剂。虽然直接的近红外光触发 PQ-ERA 光点击反应尚未实现,但该研究组引入了一种使用掺杂镧系元素的 UCNPs 的间接策略,实现了目前最快的近红外光(800 纳米、980 纳米)触发光点击反应,反应时间为 10 分钟,具有体内应用和深层组织穿透的潜力(图)。
图 9:酰基硅烷与吲哚的 PQ-ERA 光环加成和光点击反应。
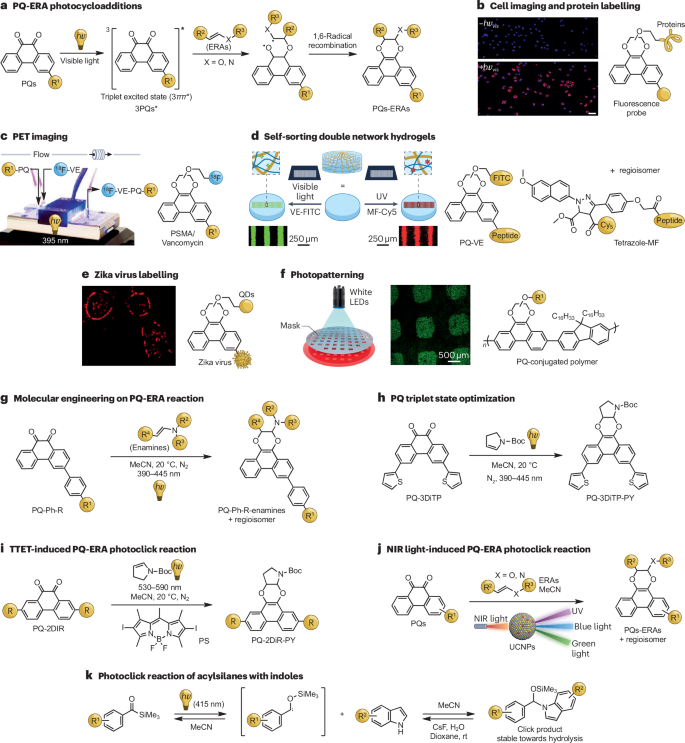 a-f,9,10-菲醌-富电子烯(PQ-ERA)光点击反应的拟议机理。可见光诱导的 PQ-ERA 反应在细胞成像和蛋白质标记(b 部分)、正电子发射断层扫描(PET)成像(c 部分)、自分选双网络水凝胶(d 部分)、寨卡病毒标记(e 部分)和光图案化(f 部分)中的应用。g,PQ-ERA 光点击反应的分子工程,以提高反应活性和选择性。h*,优化PQ三重态的性质,以前所未有的效率建立 PQ-ERA 光点击反应。i、三重-三重能量转移(TTET)诱导的PQ-ERA光点击反应。j,由上转换纳米粒子(UCNPs)辅助的近红外线(NIR)诱导的 PQ-ERA 光点击反应。k,酰基硅烷-吲哚光点击反应实例。Cy5,青绿素 5;FITC,荧光素;LED,发光二极管;MF,单富马酸盐;PS,光敏剂;PSMA,前列腺特异性膜抗原抑制剂;QDs,量子点;rt,室温;UV,紫外线;VE,乙烯基醚。b、c和f部分经参考文献许可改编。ACS。第d部分转载自参考文献。部分d转载自参考文献,施普林格自然有限公司。第e部分改编自参考文献,采用知识共享许可协议。
a-f,9,10-菲醌-富电子烯(PQ-ERA)光点击反应的拟议机理。可见光诱导的 PQ-ERA 反应在细胞成像和蛋白质标记(b 部分)、正电子发射断层扫描(PET)成像(c 部分)、自分选双网络水凝胶(d 部分)、寨卡病毒标记(e 部分)和光图案化(f 部分)中的应用。g,PQ-ERA 光点击反应的分子工程,以提高反应活性和选择性。h*,优化PQ三重态的性质,以前所未有的效率建立 PQ-ERA 光点击反应。i、三重-三重能量转移(TTET)诱导的PQ-ERA光点击反应。j,由上转换纳米粒子(UCNPs)辅助的近红外线(NIR)诱导的 PQ-ERA 光点击反应。k,酰基硅烷-吲哚光点击反应实例。Cy5,青绿素 5;FITC,荧光素;LED,发光二极管;MF,单富马酸盐;PS,光敏剂;PSMA,前列腺特异性膜抗原抑制剂;QDs,量子点;rt,室温;UV,紫外线;VE,乙烯基醚。b、c和f部分经参考文献许可改编。ACS。第d部分转载自参考文献。部分d转载自参考文献,施普林格自然有限公司。第e部分改编自参考文献,采用知识共享许可协议。
酰基硅烷与吲哚的光点击反应
在可见光(蓝光 LED)照射下,酰基硅烷中的羰基和硅基间的伸长键发生裂解,形成硅氧羰基这一活性中间体,该中间体在有机合成和材料科学领域有着广泛应用。酰基硅烷可用作可见光固化的光响应聚合引发剂,并因其硅迁移可逆性而延长了硅氧羰基在材料化学中插入 X-H 键的寿命。
2021 年,Studer 及其合作者利用酰基硅烷的吲哚衍生物开发出一种可见光介导的光点击反应(λ = 415 纳米),该反应具有产率高、动力学反应迅速、选择性强的特点,适用于连接小分子和大分子(图)。此方法成功应用于糖衍生物与吲哚生物碱的偶联、聚合物共轭、交联和折叠等方面。带有硅氧基缩醛基团的酰基硅烷在 254 纳米波长的光照下容易裂解,因此可用作吲哚的 N 保护基团。研究小组将这种方法推广到了温和条件(MeCN、420 纳米蓝光 LED)下对含有色氨酸的肽进行选择性修饰和标记,获得了极佳的产率,展现了炔吖啶点击化学的多功能性。
其他光环加成反应
除了 PQ-ERA 涉及的光点击反应之外,还有其他类型的光点击反应。例如,共轭烯(如链烯、胸腺嘧啶等)和蒽在特定条件下会发生光环加成反应。不过,这些化合物通常需要高能紫外线进行激发,进而形成共价键。另外,这些反应所形成的光产物在持续光照下往往不稳定,因此不太适合光点击化学的应用。加之这些反应底物在水中的溶解度较低,这也限制了它们在生物领域的应用。
尽管如此,一些系统已显示出向可见光诱导的光点击反应发展的潜力。例如,2018 年 Barner-Kowollik 及其同事展示了一种绿色(λ ≈ 550 nm)光诱导的反应。该体系已被用于构建水凝胶,为更广泛的可见光响应型光点击反应的发展提供了前景。
作为一种新兴的光点击反应,IV 型光点击反应自首次报道以来就备受关注。与其他光点击反应不同的是,该体系能在完全可见光控制下运行,主要包括 PQ-ERA 反应以及酰基硅烷与吲哚的光点击反应两大类。反应适用的波长范围从 390 纳米到 980 纳米。
对于 PQ-ERA 反应,已经采用了多种优化策略,包括三重态优化、TTET 和 UCNPs 技术,成功地将反应扩展至长波长可见光和近红外光。然而,目前的应用主要局限在体外环境。未来的研究应着重于利用长波长光活性成分来拓展生物应用领域。
至于酰基硅烷与吲哚的光点击反应,该系统目前仅限于 400 纳米波长范围内,主要用于聚合物化学。后续的研究工作应当致力于拓宽反应的驱动波长,并探索更多应用领域,如生物大分子标记和表面改性。
结论与展望
在过去十年里,可见光和近红外光触发的光点击反应取得了显著进展,这克服了传统体系依赖高能紫外光的局限性。这些突破为光点击反应在材料科学和生命科学的应用开辟了广阔的前景。在之前的章节中,本文探讨了几种可见光和近红外光诱导的光点击反应系统,并根据它们的潜在光激活机制进行了分类(I-IV 型)。同时,本文还重点介绍了各种复杂的设计策略,以实现每个光活性组分家族中的红移光点击反应。
设计可见光诱导的光点击体系最常用的方法是扩展已知化合物的 π 电子系统。这种方法成功应用于 II 型和 III 型光点击反应体系,目的是通过降低 HOMO-LUMO 能隙来将化合物的吸收向红光区转移,从而增强波长选择性。近期的研究报道了使用这种方法实现的蓝绿光(405-515 nm)诱导的四唑-烯环加成(NITEC)和 DASyd-烯环加成(photo-DASAC)。然而,这种方法存在一些局限性。一方面,延长 π 共轭可能导致光点击性能下降,使得实现快速且选择性的转化变得更具挑战性。另一方面,化合物在水溶液中的溶解度也会因 π 共轭的延长而降低,从而限制了它们在生物系统中的应用。
双光子或多光子吸收过程提供了一种使用能量仅为单光子转换所需能量一半或更少的光子来激发分子的方法。这些过程适用于 I、II 和 III 型光点击反应系统,展现出非线性强度依赖性,即双光子吸收(2PA)随光强的平方而增加,这为实现优异的三维空间分辨率创造了条件。这对于需要在特定体积内精确照射材料、组织或细胞的应用至关重要,如材料科学中的水凝胶形成和化学生物学中的相关应用。最近的研究显示,某些光点击前体具有可观的内在双光子吸收截面,可以直接通过双光子激发来激活。但是,这一过程需要高强度的脉冲激光(脉冲强度大于 10^6 W/cm²)。
上转换过程涉及多个光子的连续吸收,随后发射较短波长的辐射。利用这一原理,系统可以吸收多个量子的近红外光,然后将上转换激发能量转移给光点击前体。这种能量转移可以通过多种途径发生,比如紫外线或蓝光发射后光点击前体的再吸收,或者通过非三重态福斯特共振能量转移。最近的研究成功证明了这种方法在 III 型(NITEC)和 IV 型(PQ-ERA)光点击系统中的可行性。尽管近红外光诱导的点击反应极大地提升了在生物系统中应用点击化学的潜力,但仍然存在一些问题,包括上转换发光效率低、需要长时间高功率激光照射以及能量转移和再吸收效率低(UCNP → 光点击前体)。因此,未来的研究应侧重于提高 UCNP 和光点击前体之间的能量传递效率,探索共价连接的多功能系统被视为一个非常有前景的研究方向。
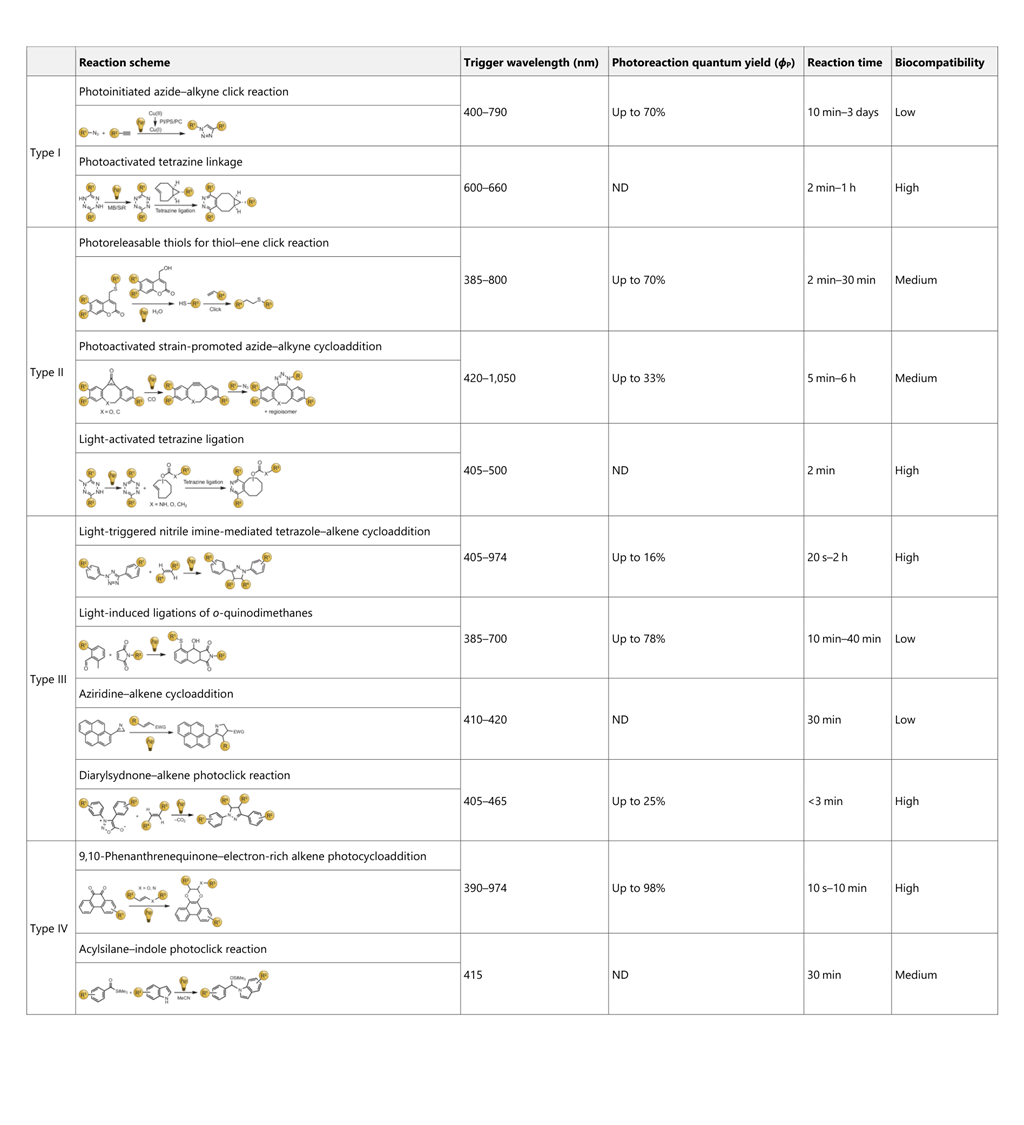
表 1 所有光点击反应类型和关键参数汇总
结论与展望
三重态-三重态能量转移(TTET)是一种有前景的替代策略,用于通过低能三重态实现红移光点击活化。这种方法特别适用于那些三重态发挥关键作用的光点击反应系统,如 NITEC 和 PQ-ERA 光点击系统。通过间接激发三重态敏化剂并随后敏化光点击元件,可以在相当红移的波长下进行光化学反应,而无需对光点击元件进行复杂的结构修改或功能化。这简化了分子设计,确保了在更长波长下的高效光操作,同时避免了新型衍生物可能出现的不可预测行为。最近使用纯有机三重态敏化剂(如 DiIbodipy 衍生物)引发绿光诱导的 PQ-ERA 光点击反应(类型 IV)的成功证明了这种方法的有效性。然而,由于 Dexter 电子交换机制,三重态增感策略可能受限于其对氧气的敏感性和距离依赖性。通过将三重态敏化光点击反应系统纳入封闭环境,如密集的聚合物薄膜、胶束和纳米通道,可以克服这些缺点。需要进一步研究此类封闭系统中的创新装配,以充分发挥其潜力。
值得注意的是,基于创新反应物结构开发新型光点击反应仍然存在大量机会,这些反应物结构允许用可见光或近红外光直接触发。尽管最近出现了 PQ-ERAs、酰基硅烷-吲哚光点击反应和光活化四嗪-反式环辛烯点击反应等成功案例,本文认为还有很多尚未探索的发展路径。例如,在开发新的可见光和近红外光诱导的光点击反应体系时,需要仔细考虑吸收光谱与光化学反应活性之间的差异。尽管这一现象在光化学中普遍存在,但对于希望将光化学反应性转移到可见光范围的研究人员来说尤为重要。Barner-Kowollik 及其同事的研究实例强调,在吸收率极低的区域中,光化学反应性往往非常显著,这表明在意外的领域可能存在浴色偏移反应性。通过致力于发现能满足日益多样化的体内应用需求的光点击分子新家族,本文可以为该领域的进一步发展铺平道路。
近期的研究主要集中在提高光点击反应的观察反应速率上。然而,与经典点击反应不同,光点击反应是通过外部传递光子来驱动的,因此,观察到的产物形成速率取决于多种因素,包括反应设置和使用的光源。因此,要优化反应本身,就必须关注影响反应效果的内部参数。值得注意的是,光反应过程中最关键的一个参数:_Φ_P(更多信息请参见表)很少被考虑。这一指标本质上衡量了光反应过程的效率。量子产率越高,表明光子在反应过程中的效率越高,从而对应用产生影响。例如,高_Φ_P 的光点击反应对体外应用非常有利。即使使用 UVA-2 范围(>340 nm)的驱动波长,由于光源强度低、曝光时间短,这些反应对细胞光毒性的风险也很小。相反,尽管使用了长波长可见光或近红外光,但_Φ_P 明显较低的光点击反应系统需要较长的曝光时间或高功率光源,可能会导致不希望出现的光毒性和热效应。
此外,有必要强调的是,本综述旨在作为一份实用指南,帮助选择合适的光点击反应进行生物应用。因此,为体外或体内使用选择合适的光点击反应系统至关重要。体外系统可使用波长在 UVA-2(340-380 纳米)和可见光范围(380-760 纳米)内的光点击反应系统,因为这些光谱范围内的光显示出最小的细胞毒性。相反,对于体内应用,建议选择驱动波长在深红光或近红外光范围内的光点击反应系统。哺乳动物组织对 650 纳米以下光线的吸收很强,导致光点击反应过程的效率降低,并可能产生光毒性。为了帮助读者比较各种类型的光点击反应,本文编制了一份概述其分类的汇总表(表)。该表评估了光点击反应的多个方面,包括反应速率、适用波长范围、生物相容性、光反应量子产率和其他相关参数。本文的目的是为读者提供更清晰的认识,使他们能够根据自己的具体要求确定最合适的方法。
总而言之,设计长波长驱动的光点击反应需要全面了解有机化学、光化学、计算化学、激光技术以及与生物系统的生物医学应用兼容性。本综述总结了当前的设计策略及其机理基础,包括直接激发(如 π 系统扩展或推拉系统)和间接激发方法(如三重态敏化、上转换纳米粒子、多光子吸收、光催化、光引发和光笼式方法)。尽管取得了这些进展,本文认为红光和近红外光驱动的光点击反应仍有很大的进一步探索和优化空间。实现这一目标是一个诱人的挑战,因为它有可能在需要精确时空控制分子活性的体内研究和新型生物正交技术开发方面做出突破性贡献。本文相信,这篇综述将在不久的将来激发人们对这些新兴的红移可光点击分子类别日益增长的兴趣,并激发多学科研究和工业创新的新思路。本文热切期待这一领域在未来几年取得令人振奋的进展。