【JACS】“一分子双靶向,抗癌力提升82%:23f化合物在AML治疗中的突破性研究”
文章标题: Discovery of Novel and Highly Potent Wee1 and HDAC Dual-Target Inhibitors for the Treatment of Acute Myeloid Leukemia 通讯作者: Hongfeng Gu, Hui Huang, Jinsong Han 文章链接: https://doi.org/10.1021/acs.jmedchem.5c01789
文章概要
急性髓性白血病(AML)是一种侵袭性极强的血液肿瘤,尽管近年来治疗手段不断进步,但靶向药物耐药性、化疗毒性及副作用仍是临床治疗的重大挑战。本研究以“合成致死”策略为基础,提出通过同时抑制Wee1激酶与组蛋白去乙酰化酶(HDAC)来增强抗癌效果,并成功设计出首个具有双靶点活性的候选小分子——化合物23f。
研究背景与设计思路
Wee1是一种丝氨酸/苏氨酸激酶,调控细胞周期G2/M及S期检查点,能诱导DNA损伤细胞进入有丝分裂,导致细胞凋亡。AZD1775是首个进入临床的Wee1抑制剂,但因剂量限制毒性而终止开发。研究发现,AZD1775单药治疗会激活CHK1通路,削弱其抗增殖效果。而HDAC抑制剂可下调CHK1表达,提示二者联用具有协同抗癌潜力。
为克服联合治疗的药代动力学复杂性与成本问题,研究团队采用药效团组合策略,将Wee1抑制剂的核心结构与HDAC抑制剂的锌结合基团(ZBG)连接,设计出一系列双靶向分子。其中,23f通过在Wee1抑制剂的甲基哌嗪位点引入HDAC药效团,兼顾两者活性。
化合物筛选与优化
研究共合成了数十种候选分子,系统评估其对Wee1与HDAC1的酶抑制活性。结果显示:
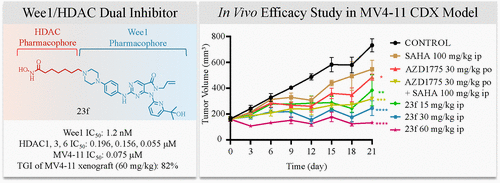
- 链长为6的连接基团(23f)在Wee1和HDAC1活性上均表现优异(IC₅₀分别为1.2 nM和0.196 μM)。
- 23f在MV4–11细胞中展现出最强的抗增殖活性(IC₅₀ = 0.076 μM),比AZD1775强约3倍,比SAHA强约8倍。
- 进一步结构优化表明,HDAC部分的“帽子基团”电子密度对活性影响显著,氯代取代在Wee1结合口袋中更具空间适配性。
机制验证与体内疗效
分子对接与CETSA实验确认23f能稳定结合Wee1与HDAC1蛋白。Western blot结果显示,23f可:
- 下调CDK1磷酸化水平(Wee1下游标志物)
- 上调组蛋白H3乙酰化水平(HDAC下游标志物)
- 激活γH2A.X表达,提示DNA损伤增强
- 抑制Wee1诱导的CHK1补偿性激活,验证其协同机制
在MV4–11小鼠异种移植模型中,23f以60 mg/kg剂量经腹腔注射,肿瘤生长抑制率(TGI)高达82%,显著优于AZD1775(29%)、SAHA(37%)及其联合治疗(58%)。同时,23f未引起体重下降或器官毒性,血液生化指标(ALT、AST、BUN)正常,显示良好安全性。
药代动力学与选择性分析
在大鼠体内,23f经腹腔注射后吸收迅速,生物利用度达157.58%,半衰期为3.63小时,优于口服途径(生物利用度仅2.21%)。HDAC亚型选择性实验显示,23f对HDAC1、HDAC3、HDAC6具有较强抑制活性,与SAHA类似,表明其为广谱HDAC抑制剂。
结论与展望
本研究首次报道了Wee1/HDAC双靶点小分子抑制剂的设计与验证,23f不仅在体内外展现出卓越的抗AML活性,还通过系统的结构优化明确了影响双靶点活性的关键因素。该分子可作为进一步临床转化的先导化合物,也为多靶点抗癌药物的开发提供了重要参考。
这项工作展示了药效团组合策略在抗癌药物设计中的巨大潜力,为AML治疗开辟了新路径。未来研究可围绕23f进行结构优化、临床前安全性评估及机制深入探讨,推动其向临床应用迈进。