【JACS】硫辅助光催化:开启氧化还原惰性底物烷基自由基生成的新纪元
文章标题: Visible-Light-Induced C–S Bond Cleavage Enables Alkyl Radical Generation from Redox-Inert Substrates
通讯作者: Sho Murakami, Hirohisa Ohmiya
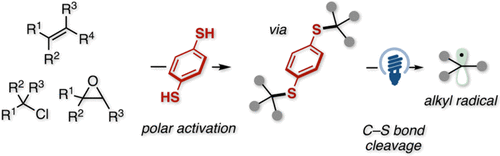
文章概要
本研究开发了一个可见光驱动的烷基自由基生成平台,其核心优势在于无需外部光催化剂或化学计量还原剂。该技术通过极性活化形成的含硫中间体进行直接光激发,利用1,4-苯二硫醇衍生物作为光活性和还原物种,实现了C-S键的均裂。这一策略不仅适用于从简单烯烃衍生的硫醚,还能成功激活烷基氯代物和环氧化合物等极难被单电子还原的氧化还原惰性底物。生成的烷基自由基可直接用于碳-碳键成键反应,为金属参与和传统光氧化还原催化提供了强有力的绿色替代方案。
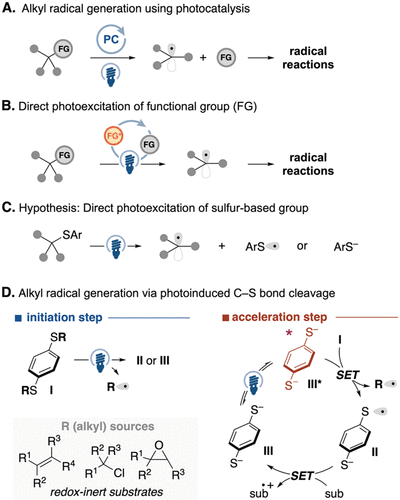
Scheme 1. Alkyl Radical Generation from Redox-Inert Substrates
引言
碳中心自由基是现代有机合成中不可或缺的中间体,但在温和条件下从易得的原料中产生烷基自由基仍具挑战,因为许多常见前体具有氧化还原惰性。传统的活化方法往往依赖贵金属催化剂、外部光催化剂或过量的牺牲试剂,这在功能团兼容性、环境可持续性和放大生产方面存在局限。尽管EDA复合物或底物直接激发有所进展,但通常仅限于特定的分子结构。京都大学的Hirohisa Ohmiya教授团队另辟蹊径,利用硫醚在极性活化下易于形成的特性,将硫原子作为光活性的“天线”,通过光激发C-S键均裂,实现了烷基自由基生成的去偶联策略,从根本上解决了惰性底物难以激活的难题。
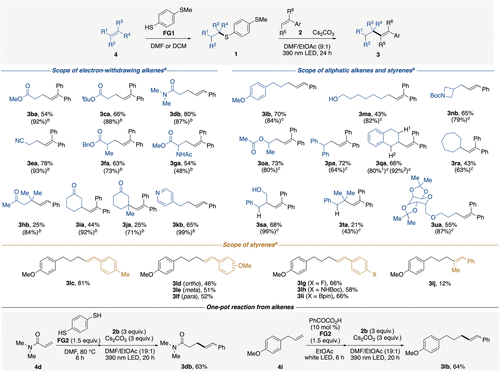
Figure 1. Substrate scope of alkene–styrene coupling. _a_Alkenylation was carried out with 1 (0.10 mmol), 2 (0.30 mmol), and Cs2CO3 (0.15 mmol) in DMF/EtOAc (0.9/0.1 mL) under 390 nm blue LED irradiation for 24 h. Isolated yields refer to the second-step alkenylation. Values in parentheses indicate the isolated yields of the activation step using FG1. _b_Activation of electron-withdrawing alkenes was carried out with 4 (0.3 mmol) and FG1 (0.33 mmol) in DMF (3 mL) at 60 °C for 6 h. _c_Activation of aliphatic alkenes was carried out with 4 (0.25 mmol), FG1 (0.75 mmol), and benzoylformic acid (0.025 mmol) in DCM (0.1 mL) under white LED irradiation for 20 h.
主要实验及结论
在反应条件筛选中,研究人员以1,4-二(烷基硫代)苯与苯乙烯的反应作为模板。实验发现,在390 nm可见光照射和碳酸铯碱性条件下,无需任何外部催化剂即可获得高达76%的产率。机理研究表明,电子丰富的硫醇盐物种在激发态下具有显著增强的还原能力。为了验证普适性,团队考察了多种烯烃前体,发现无论是电子欠缺的丙烯酸酯还是结构简单的脂肪族烯烃,通过硫-烯(thiol-ene)点击反应转化成的硫醚中间体,都能在光照下高效生成烷基自由基并与苯乙烯偶联。
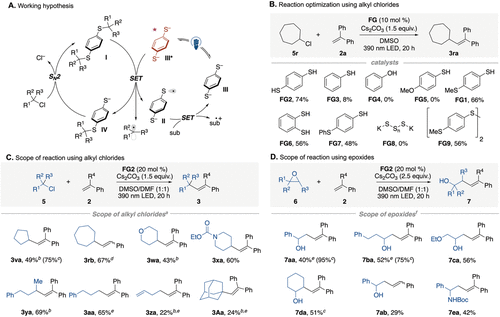
Figure 2. Substrate scope of catalytic generation of carbon radicals from alkyl chlorides and epoxides. _a_Reaction was carried out with 5 (0.10 mmol), 2 (0.30 mmol), FG2 (20 mol %), and Cs2CO3 (0.15 mmol) in DMSO/DMF (0.5/0.5 mL) under blue LED (390 nm) irradiation for 24 h. _b_DMSO was used. _c_NMR yield. _d_10 mol % FG2 was used. _e_50 mol % of FG2 was used. _f_Reaction was carried out with 6 (0.10 mmol), 2 (0.30 mmol), FG2 (20 mol %), and Cs2CO3 (0.25 mmol) in DMSO/DMF (0.5/0.5 mL) under blue LED (390 nm) irradiation for 20 h.
更为突破性的进展在于对烷基氯代物和环氧化合物的催化活化。烷基氯因其极高的还原电位,通常被认为难以通过单电子转移机制激活。本研究证明,使用催化量的1,4-苯二硫醇,烷基氯可以通过S~N~2亲核取代先原位转化为硫醚,随后受光激发发生C-S键均裂,顺利转化为偶联产物。同样,环氧化合物也能通过硫亲核试剂的区域选择性开环形成硫醚中间体,从而开启自由基反应途径。DFT计算进一步证实,激发态的硫醇负离子通过系际交叉进入三重激发态,仅需较低的活化能即可完成C-S键断裂,生成稳定的烷基自由基。
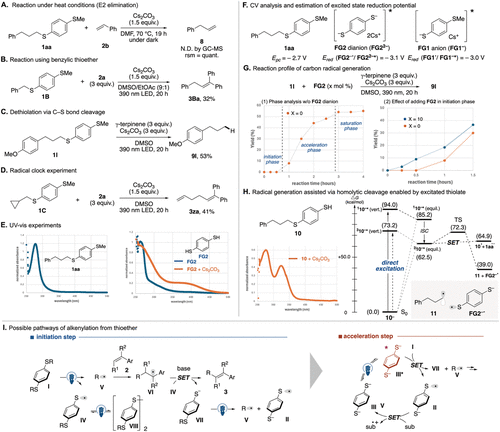
Figure 3. Mechanistic experiments and possible pathways.
总结及展望
该研究成功构建了一个基于硫辅助光活化的通用平台,彻底摆脱了对外部金属催化剂和昂贵光催化剂的依赖。这一策略通过极性成键与自由基断裂的巧妙结合,使原本性质稳定的烷基氯和环氧化合物在可见光下焕发活力。这种方法不仅操作简便、区域选择性高,而且具有极佳的原子经济性,为药物分子修饰和复杂分子合成提供了高效且低碳的新工具。未来,这种利用原位生成的有机中间体捕获光能的策略,有望在更多具有挑战性的化学转化中发挥巨大潜力。