【Angew.Chem】23种取代基如何精准调控光开关寿命?——深度解析芳基偶氮吡唑热稳定性机制
芳基偶氮吡唑(arylazopyrazole)作为新一代光开关分子,因其优异的光致变色性能和可调控的热稳定性,在光药物、智能材料和分子存储等领域展现出巨大潜力。本文基于《Angewandte Chemie》最新研究成果,系统总结了23种对位取代基对芳基偶氮吡唑热半衰期的影响机制,揭示了电子效应如何决定其热异构化路径,并提出了可预测的分子设计策略。
🔬研究背景:为何芳基偶氮吡唑备受关注?
传统的光开关分子如偶氮苯虽具有高光稳定性和合成简便性,但存在两个关键缺陷:
- E/Z异构化不完全;
- 在室温下热回转速度过快,Z异构体寿命仅为几天。
相比之下,芳基偶氮吡唑不仅具备近乎完全的双向光开关能力,还能通过结构修饰实现从几分钟到近1000天的热半衰期调控。尤其是对位取代基的调控作用,成为实现精确设计的关键。
🧪合成策略:构建23种取代芳基偶氮吡唑库
研究团队以芳基偶氮-1,3,5-三甲基吡唑为母体,设计并合成了23种不同对位取代的衍生物。合成路线主要包括: 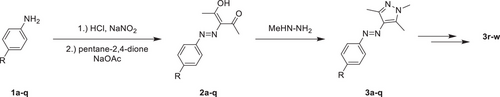
- 对位取代苯胺经重氮化生成中间体;
- 与甲基肼缩合得到目标芳基偶氮吡唑。
大多数化合物合成收率在66%–92%之间,部分通过官能团转换进一步扩展结构多样性,如还原、甲基化、酰胺化等。
🌈光谱特性:吸收峰如何响应取代基变化?
所有化合物在DMSO中进行UV/Vis光谱测试,发现: 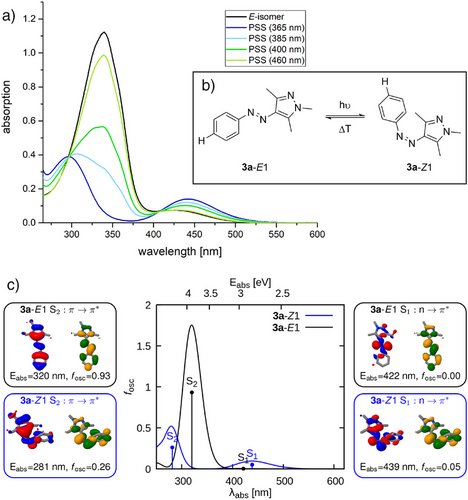
- 绝大多数化合物在365 nm照射下可实现 ≥94% 的Z异构体含量;
- 强电子给体(如–NEt₂、–OH)导致π–π*吸收峰显著红移,最长达60 nm;
- 强电子吸收基(如–NO₂、–CN)也引起红移,但程度较轻。
这种吸收峰的变化不仅影响光致异构化效率,也反映了分子轨道能级的变化趋势。
⏳热半衰期:从数天到数秒,取代基如何决定稳定性?
实验测得的热半衰期跨度极大: 
- 中性或弱电子效应取代基(如–F、–Cl、–Me)保持较长半衰期(约10天);
- 强电子吸收基(如–NO₂)或强电子给体(如–OH)导致半衰期骤降至秒级。
这表明无论是电子给体还是吸收基,都会加速Z→E热回转过程,但机制却截然不同。
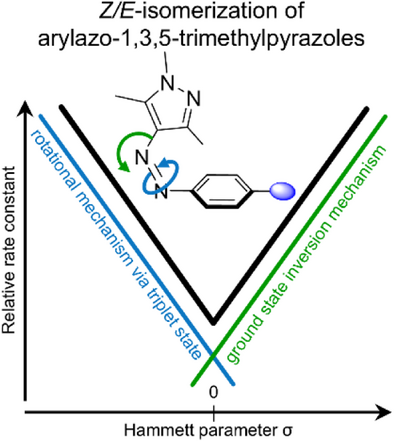
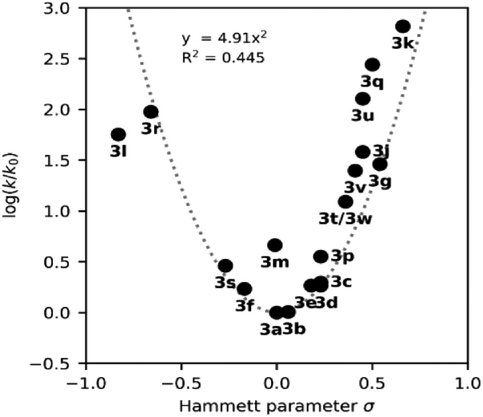
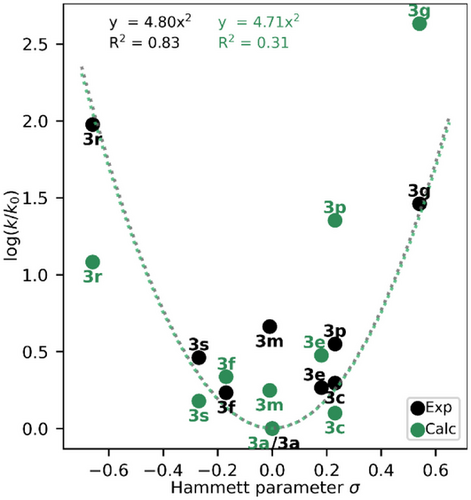
📈哈梅特分析:σ值揭示机制转换点
通过绘制哈梅特图(Hammett plot),研究发现: 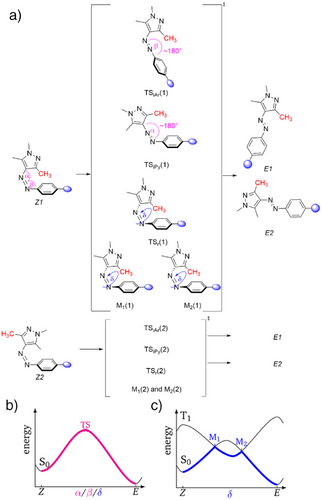
- 热回转速率与σ值呈现典型V型曲线;
- σ值越偏离0,热回转越快;
- 中间σ值(如–H、–Ph)对应最长半衰期。
这一趋势暗示不同电子效应引发不同的热异构化机制。 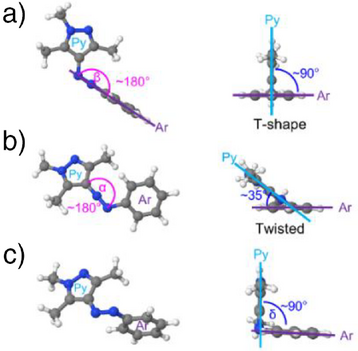
⚙️机制解析:两大路径主导热回转过程
研究采用密度泛函理论(DFT)和非绝热过渡态理论(NA-TST)计算,揭示两种主要机制: 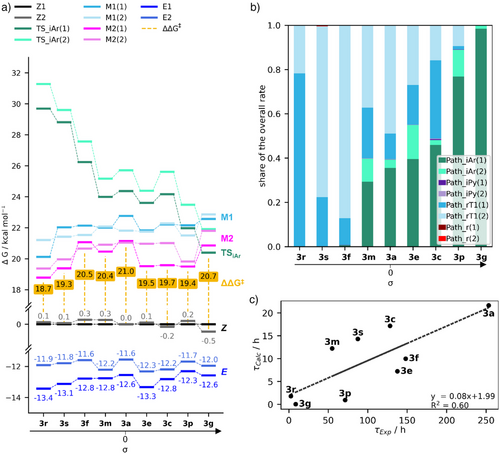
1. 单重态反应路径(Path iAr)
- 适用于强电子吸收基;
- 通过芳环邻近氮原子的平面内反转;
- 过渡态呈“T型”结构,芳环与吡唑垂直排列。
2. 三重态旋转路径(Path rT1)
- 适用于强电子给体;
- 通过非绝热跃迁进入最低三重态;
- 旋转异构化绕N=N键进行,伴随电子离域增强。
中性取代基则呈现两种机制混合,导致热稳定性增强。
🧠电子结构分析:为何电子效应决定路径选择?
- Mulliken电荷分析显示,电子给体增强N=N键的π共轭,稳定三重态;
- Wiberg键指数表明,电子给体提高偶氮键离域性;
- 相反,电子吸收基降低共轭性,提升单重态反应路径的活化能。
因此,电子效应不仅影响轨道能级,还决定了反应路径的能垒高低。
📊计算与实验对比:预测模型的准确性如何?
尽管计算半衰期普遍偏短(约为实验值的1/12.5),但趋势吻合度高:
- 计算与实验哈梅特图均呈V型;
- 排除三重态机制后,拟合曲线变为线性,误差显著增加;
- 说明三重态机制在解释电子给体效应中不可或缺。
🎯结语:如何实现光开关的精准设计?
本研究首次系统揭示了对位取代基如何通过电子效应调控芳基偶氮吡唑的热稳定性,提出了如下设计原则:
- 若需长寿命Z异构体,应选择中性或弱电子效应取代基;
- 若需快速回转,应使用强电子吸收或给体;
- 结合哈梅特参数与计算模型,可实现热半衰期的定量预测。
这一机制洞察为光开关分子的理性设计提供了坚实基础,助力其在生物医学与智能材料领域的广泛应用。